Hyperaldosteronism: Practice Essentials, Pathophysiology, Etiology
Maybe your like
Normal aldosterone physiology
Aldosterone participates in homeostasis of circulating blood volume and serum potassium concentration via a feedback loop involving the zona glomerulosa of the adrenal cortex.
Mechanisms of aldosterone secretion include the following:
- Stimulants - Secretion is stimulated by actual or apparent blood volume depletion (detected by stretch receptors), increased potassium ion concentrations, and angiotensin II
- Inhibitors - Production is suppressed by hypervolemia, hypokalemia, atrial natriuretic peptide (ANP), and dopamine
- Acute regulation - ACTH stimulates secretion in an acute, transient fashion but does not appear to play a significant role in long-term regulation
The renin-angiotensin-aldosterone (R-A-A) axis involves multi-organ coordination. Prorenin synthesis and conversion to renin are stimulated by volume contraction, beta-adrenergic activity, and prostaglandins I2 and E2. Renin converts hepatic angiotensinogen to angiotensin I, which angiotensin-converting enzyme (ACE) converts to the octapeptide angiotensin II in the lungs. Angiotensin II and its metabolite, angiotensin III, both stimulate aldosterone secretion.
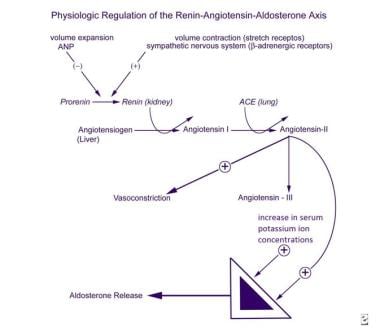 Physiologic regulation of the renin-angiotensin-aldosterone (R-A-A) axis. View Media Gallery
Physiologic regulation of the renin-angiotensin-aldosterone (R-A-A) axis. View Media Gallery Cellular and genetic factors
Metoclopramide has been shown to increase aldosterone secretion, suggesting that dopamine may tonically inhibit aldosterone release. The physiologic roles of adrenomedullin and vasoactive intestinal peptide (VIP) remain to be clarified, although both are produced in the rat zona glomerulosa.
Intracellular ionized calcium concentration is required for the normal function of stretch receptors and prostaglandin I2 and E2 synthesis and secretion.
Aldosterone is synthesized from cholesterol in six steps; the final two steps are unique to aldosterone. Aldosterone synthase, encoded by CYP11B2 on chromosome 8q24.3-tel, catalyzes aldosterone synthesis. [4]
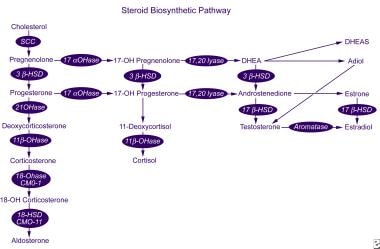 Steroid biosynthetic pathway. View Media Gallery
Steroid biosynthetic pathway. View Media Gallery Aldosterone action on target tissues (eg, the distal renal tubule, sweat glands, salivary glands, and epithelium of the large intestine) is mediated via a specific mineralocorticoid receptor. Mineralocorticoid receptors exhibit equal affinity for mineralocorticoids and cortisol, yet the aldosterone receptors in the distal tubule and elsewhere are protected from cortisol-mediated activation by 11β-hydroxysteroid dehydrogenase type 2, which locally converts cortisol to inactive cortisone.
Primary aldosteronism
The term primary hyperaldosteronism (or primary aldosteronism [PA]) refers to a renin-independent increase in the secretion of aldosterone. This condition is principally a disease of adulthood, with its peak incidence in the fourth to sixth decades of life.
More than 90% of cases of PA are due either to an APA, which accounts for around 35% of cases (30-40%), or to idiopathic hyperaldosteronism (IHA), which accounts for around 60% of cases (almost all of which are bilateral). Unilateral adrenal hyperplasia is a rare cause of PA, accounting for 1-2% of cases. About 1% of patients present with adrenocortical carcinomas (ACCs) that are purely aldosterone-secreting and are usually large at the time of diagnosis; 1% present with familial hyperaldosteronism, and 1% present with an ectopic APA or carcinoma. [5]
Unilateral adrenal hyperplasia accounts for 14-17% of all cases of unilateral PA. The prevalence of cortical adenoma within cortical hyperplasia is estimated to be 6-24%. The clinical presentation and outcome of patients with unilateral primary hyperaldosteronism are similar regardless of the histopathologic diagnosis. Unilateral adrenocortical hyperplasia is rare. [6]
APAs (sometimes referred to as aldosteronomas) are usually benign, encapsulated adenomas that are less than 2 cm in diameter. Most cases are solitary, although in as many as one third of cases, evidence exists of nodularity in the same adrenal gland, suggesting that the condition has arisen in a previously hyperplastic gland.
Patients with IHA have bilateral thickening and variable nodularity of their adrenal cortex. A wide spectrum of severity exists for this disorder, which may go undetected for long periods with no hypokalemia and only mild hypertension. It has been suggested that IHA arises as a result of an undetected adrenal cortex–stimulating factor. Alternatively, the disorder may arise as a result of an activating mutation in an adrenal cortex–specific gene. Neither hypothesis has been proven.
Inherited forms of primary hyperaldosteronism account for only 1% of cases but are more likely to occur during childhood years. These forms include familial hyperaldosteronism (FH) types I, II, and III.
Familial hyperaldosteronism type I
FH-I, or GRA, may be detected in asymptomatic persons during screening of the offspring of affected individuals, or patients may present in infancy with hypertension, weakness, and failure to thrive due to hypokalemia. FH-I is inherited in an autosomal dominant manner and has a low frequency of new mutations.
FH-I arises as a result of unequal crossing over of highly related CYP11B1 (the 11β-hydroxylase gene) and CYP11B2 (the aldosterone synthase gene) during meiosis, producing an anti-Lepore-type fusion product. [7, 8] This genetic rearrangement causes the expression of CYP11B2 to be placed under the control of the CYP11B1 promoter and the aldosterone synthesis to be abnormally regulated by ACTH rather than by the renin-angiotensin system.
The result is ACTH-dependent aldosterone production; in addition, there is production of 17-hydroxylated analogues of 18-hydroxycortisol, which is under ACTH regulation from ectopic enzyme expression in the zona fasciculata. Bilateral hyperplasia of the zona fasciculata occurs, and high levels of novel 18-hydroxysteroids appear in the urine. Adenoma formation is rare, but patients do have a significant increase in incidence of cerebrovascular aneurysms, for which they require screening.
Familial hyperaldosteronism type II
FH-II is a non–glucocorticoid-suppressible, inherited form of hyperaldosteronism. Like FH-I, it is inherited in an autosomal dominant manner. In contrast to FH-I, some FH-II kindreds exhibit a high rate of adenoma formation.
The mechanism and gene locus have not yet been identified, though CYP11B and the renin and angiotensin II receptor genes have been excluded. However, linkage has been established for a number of families to band 7p22. [9, 10] It has also been speculated that FH-II is not a single disorder.
Familial hyperaldosteronism type III
FH-III is a rare autosomal dominant form of PA characterized by early onset hypertension, nonglucocorticoid-remediable hyperaldosteronism, and hypokalemia. Germline heterozygous missense mutations of the KCNJ5 gene, encoding Kir3.4, a member of the inwardly rectifying K+ channel family, have been identified as a cause of FH-III. [11, 12, 13]
The clinical phenotype of patients harboring the above mutations ranges from severe PA and hypertension refractory to medical treatment that requires bilateral adrenalectomy, to mild or moderate hypertension responsive to medical therapy. In some patients, adrenal hyperplasia has been described.
Various studies from different centers report a prevalence of somatic KCNJ5 mutations in sporadic APAs, ranging from 30-65%. [11, 14, 15, 16]
APAs with KCNJ5 mutations are more prevalent in females than males and in younger patients. They are also associated with higher preoperative aldosterone levels. They are not related with the tumor size, but they are related with higher aldosterone levels and lower K+ concentrations.
Transcriptome and real-time polymerase chain reaction (PCR) analyses demonstrate that APAs with KCNJ5 mutations exhibit increased expression of the CYP11B2 gene and its transcriptional regulator NR4A2, thus increasing aldosterone production. It has also been found that APAs with and without KCNJ5 mutations display slightly different gene expression patterns. [16] Another study reports KCNJ5 messenger RNA (mRNA) levels to be higher in the APAs with KCNJ5 mutations and significantly higher in APAs than in cortisol-producing adenomas and pheochromocytomas. [15]
Somatic mutations in ATP1A1 (gene that encodes the alpha-1 [catalytic] subunit of the Na+/K+ adenosine triphosphatase [ATPase], a member of the P-type ATPase family), ATP2B3 (gene that encodes the plasma membrane calcium-transporting ATPase 3 [PMAC3], another member of the P-type ATPase family), or CACNA1D (gene that encodes CaV1.3, the alpha subunit of an L-type voltage-gated calcium channel) are present in approximately 6%, 1% and 8% of all cases of an APA, respectively. [18]
Secondary hyperaldosteronism
Secondary hyperaldosteronism is a collective term for a diverse group of disorders characterized by physiologic activation of the R-A-A axis as a homeostatic mechanism designed to maintain serum electrolyte concentrations or fluid volume. In the presence of normal renal function, it may lead to hypokalemia.
Secondary hyperaldosteronism can be divided into two categories, one with associated hypertension and one without. The former category includes renovascular hypertension, which results from renal ischemia and hypoperfusion leading to activation of the R-A-A axis. The most common causes of renal artery stenosis in children are fibromuscular hyperplasia and neurofibromatosis. Hypokalemia may occur in as many as 20% of patients.
PRA levels are often in the reference range, but elevated levels of PRA may be detected after provocation with a single dose of captopril 1 mg/kg. Renal ischemia is also thought to underlie the secondary hyperaldosteronism observed in malignant hypertension.
Hyperreninemia and secondary aldosteronism have also been reported in patients with pheochromocytoma, apparently as a result of functional renal artery stenosis. Renin-producing tumors are very rare, and very high levels of PRA (up to 50 ng/mL/h) are noted, frequently with an increased prorenin-to-renin ratio. The tumors are generally of renal origin and include Wilms tumors and renal cell carcinomas.
Hyperkalemia due to chronic renal failure also causes secondary hyperaldosteronism. Low sodium-to-potassium ratios can be measured in saliva and stool. Cyclosporine-induced hypertension in solid organ transplant patients may also involve a component of hyperaldosteronism.
Secondary hyperaldosteronism in the absence of hypertension occurs as a result of homeostatic attempts to maintain the sodium concentration or circulatory volume or to reduce the potassium concentration. Clinical conditions in which it may arise include diarrhea, excessive sweating, low cardiac output states, and hypoalbuminemia due to liver or renal disease or nephrotic syndrome. Secondary hyperaldosteronism may also occur developmentally in newborn infants (see below).
Increased mineralocorticoid dependency in the young
The mineralocorticoid dependency of sodium reabsorption is increased during infancy and childhood, peaking in the neonatal period before decreasing progressively with advancing age. This increase occurs because the reabsorption of sodium and water by the proximal tubule is least efficient in early life, resulting in an increased sodium and water load at the level of the distal renal tubule.
Because sodium and water resorption from the distal tubule is mediated by the R-A-A axis, the PRA is approximately 10-fold to 20-fold higher in a newborn infant than in an adult. Consequently, neonates show relative increases in aldosterone production rates (>300 µg/m2/day vs 50 µg/m2/day in an adult) and plasma aldosterone concentrations (80 pg/dL vs 16 pg/dL). These increases in early life explain why young infants exhibit profound clinical symptoms of hyperaldosteronism that gradually improve with advancing age.
Tag » Why Does Aldosterone Decrease Potassium
-
Physiology, Aldosterone - StatPearls - NCBI Bookshelf
-
[Effects Of Potassium On Renin And Aldosterone] - PubMed
-
Aldosterone Raises Blood Pressure And Lowers Potassium (video)
-
Aldosterone And Potassium Secretion By The Cortical Collecting Duct
-
Extrarenal Effects Of Aldosterone On Potassium Homeostasis
-
Aldosterone | You And Your Hormones From The Society For ...
-
Aldosterone - Wikipedia
-
Aldosterone: What It Is, Function & Levels - Cleveland Clinic
-
Hyperaldosteronism: What It Is, Causes, Symptoms & Treatment
-
Effect Of Increased Potassium Intake On The Renin–angiotensin ...
-
Hypokalemia And Pendrin Induction By Aldosterone - AHA Journals
-
Relation Between Potassium Balance And Aldosterone Secretion In ...
-
Aldosterone: Sodium And Potassium Balance - Na+/K+ ... - YouTube
-
A Proposed Cybernetic System For Sodium And Potassium Homeostasis