Structure Of A Mitochondrial ATP Synthase With Bound Native ... - ELife
Maybe your like
All known mitochondrial ATP synthases form dimers in the membrane through transmembrane Fo subunits. However, in E. gracilis, only subunit c and a set of phylum-specific subunits were identified (Perez et al., 2014; Yadav et al., 2017; Zíková et al., 2009), suggesting a divergent Fo composition. The core subunit a mediates proton translocation and contains the strictly conserved R176 (Saccharomyces cerevisiae numbering). Despite its functional importance, subunit a was identified neither in the E. gracilis genome project (Ebenezer et al., 2019), nor in the mitochondrial genome and transcriptome analysis (Dobáková et al., 2015). Our cryo-EM map allowed tracing of subunit a, for which we identified six structurally conserved membrane-embedded helices (H1a to H6a) directly from side-chain densities (Figure 2—figure supplement 1A to C). Using this information, we then found the matching sequence in the available genomic data and mapped it to a mitochondrial contig, which also contains subunit 8 and nad1 in a single open reading frame (Figure 2—figure supplement 1E). Thus, by combining the information from cryo-EM and sequencing, we report the most divergent subunit a found up to date.
In all previously reported ATP synthase structures, the horizontal helix H5a bends around the c-ring, following its curvature, thereby contacting four of the ten subunits in the c-rings of yeast and algae (Figure 2B,D) (Allegretti et al., 2015; Guo et al., 2017; Hahn et al., 2016). By contrast, in the E. gracilis structure, the N-terminal part of H5a is kinked towards the lumen, and therefore does not interact with the c-ring, and instead extends towards the lumenal membrane surface (Figure 2A,C). This structural rearrangement results in a smaller interface between subunits a and c, with only three c-subunits forming interactions, which also has implications for the formation of the matrix half-channel, as discussed below.
Figure 2 with 1 supplement see all Download asset Open asset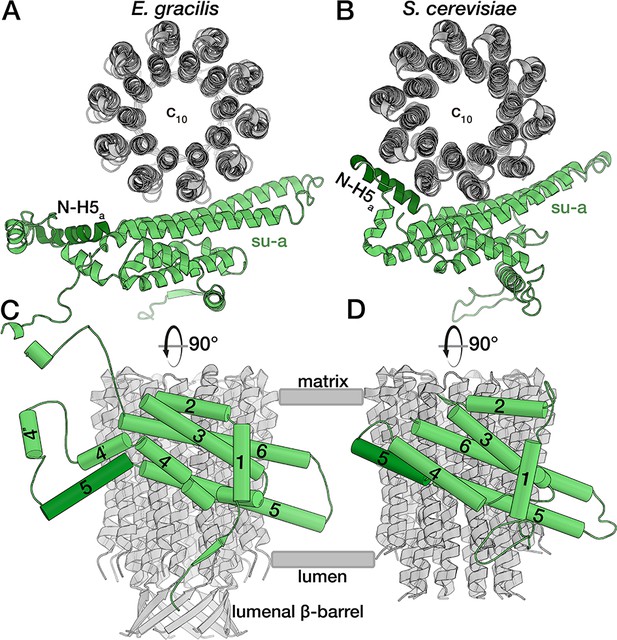
E. gracilis subunit a fold comparison.
Top view (upper panel) and side view (lower panel) of the E. gracilis (left) and S. cerevisiae (right) (Guo et al., 2017) subunit a (green) and c-ring (grey). Both structures contain the conserved H1-6a, with E. gracilis displaying two helices (H4’a and H4’’a) in an extension segment and a C-terminal extension. Whereas the N-terminal region of H5a (dark green) is kinked towards the c-ring in the yeast complex, it extends towards the lumen in the E. gracilis structure, thereby diminishing its interface with the c-ring. Unlike its yeast homolog, the N-terminus of E. gracilis subunit a is not involved in dimerisation, but contributes a strand to a β-sheet along the lumenal side of the detergent micelle.
In the yeast mitochondrial ATP synthase, subunit a interacts with transmembrane subunits b, f, i/j, k, 8 and membrane-associated subunit d (Guo et al., 2017). Since no homologs were reported for any of these subunits in E. gracilis or T. brucei (Perez et al., 2014; Yadav et al., 2017; Zíková et al., 2009), we next investigated their putative location through superimposition of our Fo model with the yeast counterpart (Guo et al., 2017). Based on the matching position and topology of the transmembrane helices as well as conserved positions of interactions with subunit a, we identified all six associated subunits, which are structurally conserved, but display no significant sequence similarity to yeast counterparts (Figure 3). Subunits b, f, i/j, k and 8 contain a single transmembrane helix associated with subunit a, whereas subunit d forms a clamp around subunit 8 at the base of the peripheral stalk, containing a structurally conserved two-helix motif at the matrix side of the membrane (Figure 3A). Finally, subunit k is bound peripherally to subunit a, as in yeast, however the H5a kink results in a 15-Å displacement of the transmembrane helix of subunit k away from the c-ring, compared to its yeast counterpart (Figure 3B). These data show that despite sequence divergence, the assembly of the central Fo subunits around subunit a is architecturally conserved between Euglenozoa and Metazoa.
Figure 3 Download asset Open asset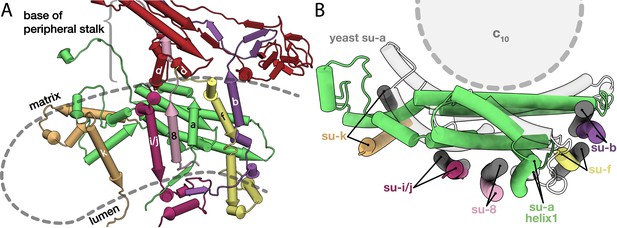
Conserved subunits of the Fo region.
(A) Side view of the conserved E. gracilis Fo subunits. Transmembrane helices with structural equivalents in yeast are labelled. (B) Top view of the superimposed conserved Fo subcomplexes from E. gracilis (coloured) and yeast (grey) PDB ID: 6B2Z (Guo et al., 2017). Although subunit k does not superimpose well, it occupies the same position relative to the H5a.
The striking architectural divergence of the E. gracilis ATP synthase dimer is brought about by euglenozoa-specific subunits and extensions of the structurally conserved Fo subunits, which render them on average 2.5 times larger than in the yeast mitochondrial ATP synthase. Only subunit b is truncated. The extensions of the conserved Fo subunits are mostly involved in forming interactions with the euglenozoa-specific subunits, thus providing a platform for the observed increased molecular mass of the Fo (Figure 4—figure supplement 1A,C).
The additional 13 euglenozoa-specific Fo subunits determine the architecture of the ATP synthase dimer, giving the Fo a markedly different overall shape, making it almost three times the size of its yeast counterpart (Figure 4—figure supplement 1). They contribute to the dimerization interface, the peripheral stalk and Fo periphery. The C-terminal helix of euglenozoa-specific ATPEG1 (H5EG1) extends 50 Å from the membrane region into the lumen, where it interacts with the N-terminal extensions of subunit c, which together form a ten-stranded β-barrel (Figure 1C and D) protruding ~20 Å into the lumen. The N-terminal residues of subunit c (A24, I25) form a hydrophobic interface with hydrophobic residues (M148, M152, L155, I159, L163) of the amphipathic ATPEG1 helix. The position of the lumenal H5EG1 on the c-ring β-barrel remains largely unchanged in all three rotational states, suggesting a mechanism of transient rotor-stator interaction during c-ring rotation. A similar lumenal interaction has previously been reported in the bovine ATP synthase, where subunit e extends from the membrane to contact the c-ring (Zhou et al., 2015). In the porcine ATP synthase tetramer, subunit e has been proposed to interact with the 6.8 kDa proteolipid, which has been suggested to reside inside the c-ring (Gu et al., 2019).
Other euglenozoa-specific Fo subunits contain structural domains that were shown to be functionally important in mitochondria (Figure 1E to G). ATPTB1 is a membrane-associated protein on the matrix side of the Fo periphery that adopts an Mdm38-like fold, which was shown to associate with yeast mitochondrial ribosomes at the inner mitochondrial membrane (Frazier et al., 2006). ATPTB3 is an isocitrate dehydrogenase ortholog that adopts a Rossman fold located at the tip of the peripheral stalk. ATPEG5 is a structurally conserved ortholog of the cytochrome c oxidase subunit VIb superfamily.
Tag » Where Is Atp Synthase Located In The Mitochondrion
-
Mitochondrial ATP Synthase: Architecture, Function And Pathology
-
Optimization Of ATP Synthase Function In Mitochondria ... - Frontiers
-
ATP Synthase - An Overview | ScienceDirect Topics
-
Mitochondria, Cell Energy, ATP Synthase | Learn Science At Scitable
-
Where Is ATP Synthase Located In The Mitochondrion? A. Electron ...
-
ATP Synthase - Wikipedia
-
Assembling The Mitochondrial ATP Synthase - PNAS
-
ATP Synthase: Structure, Function And Inhibition - De Gruyter
-
ATP Synthase | Enzyme - Britannica
-
Structure And Function Of Mitochondrial Membrane Protein Complexes
-
ATP Synthase (video) | Cellular Respiration - Khan Academy
-
ATP Synthase, F0 Complex, Subunit F, Mitochondria, Fungi (IPR019727)
-
Mitochondria | BioNinja