Mối Liên Quan Rối Loạn điện Giải Với Rối Loạn Nhịp Tim, Các Nguyên ...
Có thể bạn quan tâm
Theo thống kê qua thực hành lâm sàng trên 200 bệnh nhân điều trị ở phòng bệnh nặng khoa Nhịp Tim học bệnh viện Nhân Dân 115 trong năm 2015, tỷ lệ các bệnh nhân có nồng độ kali máu < 3,5 mEq/l lên đến > 40% bệnh nhân, nếu tính mức nồng độ kali < 4,0 mEq/l, tỷ lệ này lên đến > 50% các bệnh nhân nặng.
TS Phạm Hữu Văn
Điều này cũng phù hợp với các thống kê được thông báo trong “Các hướng dẫn mới cho thay thế kali trong thực hành lâm sàng” của Hội Y học Hoa Kỳ năm 2000. Do tầm quan trọng to lớn về vai trò điện giải trong thực hành lâm sàng, đặc biệt trong thực hành tim mạch, chúng ta cần xem xét kỹ lại vấn đề này.
Trong khi một số điện giải đóng một vai trò trong sự hình thành điện thế hoạt động qua màng tế bảo (action potential: AP), những thay đổi trong điện thế hoạt động liên quan rõ ràng nhất đến rối loạn nhịp tim phụ thuộc lớn vào mức độ kali. Độ chênh lệch (gradient) kali là một yếu tố quyết định quan trọng nhất cho độ lớn của điện thế qua màng lúc nghỉ (transmenbrane resting potential: TRP), thứ hai, tần số tăng lên của điện thế (dV / dt) pha 0 và do đó quyết định đến cả tốc độ dẫn truyền. Dẫn truyền qua màng tế bào đối với kali, hoặc giảm, rất có thể là yếu tố quyết định chính của quá trình khử cực chậm tự phát trong giai đoạn 4. Như vậy kali có tác dụng rõ rệt trên cả hai dẫn truyền và tính tự động. Hơn nữa, các đặc tính điện sinh lý bị thay đổi trong mức độ của kali gặp trong y học lâm sàng, một trạng thái như vậy, với ngoại lệ hiếm hơn, không được nhận thấy với các ions khác như canxi, magiê hoặc natri. Các ions sau ảnh hưởng đến khả năng hoạt động và gây rối loạn nhịp thực nghiệm ở nồng độ không sinh lý và thường không phù hợp với sự sống thông thường. Do đó, tất cả các chất điện giải, chuyển hóa kali bị rối loạn giải thích cho đa số các rối loạn nhịp lâm sàng. Đối với những lý do tương tự, ngoại trừ khả năng của natri và canxi để phục hồi ức chế dẫn truyền do kali gây ra, chỉ có kali là điện giải có các đặc tính chống loạn nhịp có ý nghĩa lâm sàng.
Kali (K), canxi (Ca), natri (Na) và magiê (Mg) đóng một vai trò trong sự hình thành rối loạn nhịp thực nghiệm. Tuy nhiên, trong các tình huống lâm sàng, thay đổi nồng độ kali chịu trách nhiệm cho phần lớn các rối loạn nhịp như vậy. Điều này là thực sự, vì trong phạm vi các mức độ của chất điện giải khác nhau gặp phải trong các rối loạn lâm sàng, kali là chất điện giải có khả năng làm thay đổi các thuộc tính điện sinh lý của tim nhất. Do đó, thảo luận của chúng ta sẽ giải quyết chủ yếu với những ảnh hưởng của nồng độ kali bị rối loạn và đề cập một cách ngắn gọn về vai trò có thể có của các chất điện giải còn lại. Nguyên nhân của hạ kali máu ở người lớn, lâm sàng và điều chỉnh cũng được trình bày một cách khái quát.
KALI
Kali và điện thế hoạt động qua màng tế bào
Phần này sẽ trình bày một cách ngắn gọn về vai trò của kali trong (1) duy trì điện thế hoạt động (action potential: AP), (2) hình thành, và (3) dẫn truyền xung động.Trong quá trình tâm trương (pha 4 của chu kỳ), màng tế bào cho phép kali thấm qua dẫn đến một sự mất kali thường xuyên giữa các tế bào. Do khả năng thấm có chọn lọc của kali, độ lớn của điện thế màng lúc nghỉ (transmembrane resting potential: TRP) phụ thuộc phần lớn vào nồng độ kali qua màng. Mối quan hệ này của TRP, kali được thể hiện trong phương trình Nernst dự đoán gia tăng kali ngoại bào làm giảm TRP (trở nên âm tính ít hơn) trong khi hạ thấp kali ngoại bào gây ra sự tăng phân cực, hoặc tăng TRP (trở nên âm tính nhiều hơn).
Tỷ lệ tăng lên (độ biên thiên điện thế: dV/dt) của pha 0, một yếu tố quyết định quan trọng của tốc độ dẫn truyền, phụ thuộc phạm vi rất lớn vào độ lớn của TRP. TRP càng lớn (âm hơn) TRP lúc bắt đầu giai đoạn 0, dV/dt của giai đoạn 0 và tốc độ dẫn truyền càng lớn. Vào bất cứ trạng thái rõ ràng nào đó của màng, mối quan hệ này của dV/dt ở giai đoạn 0 đến TRP đều thể hiện đáp ứng của màng. Kể từ khi tăng kali ngoại bào làm giảm TRP, dV/dt pha 0 sẽ bị giảm và dẫn truyền bị suy yếu ( hình 1).
Cần lưu ý ở điểm này, giảm TRP sẽ ở thời gian tương tự đưa TRP đến gần hơn điện thế ngưỡng (threshold potential: TP) và độ mạnh của kích thích đã cho cần thiết để giảm TRP đến TP (tính chịu kích thích) vì vậy sẽ giảm. Do đó, có thể suy giảm dẫn truyền do giảm TRP, trong phạm vi hẹp của tăng kali máu, có thể phải vượt qua một mức độ nào đó do sự gia tăng kích thích. Kết quả, dẫn truyền thay đổi với sự thay đổi nồng độ kali [5], đôi khi, có thể phản ánh tổng hợp biến đổi dV/dt và tính chịu kích thích.
Khử cực chậm trong pha 4, còn gọi là khử cực chậm tâm trương, thể hiện đặc tính của tế bào tự động, rất có thể do dòng đi vào từ từ của natri tại một thời điểm khi dòng đi ra của kali giảm đi hoặc bị ức chế hoàn toàn. Ức chế khử cực tâm trương do tăng kali máu được cho là do tăng dẫn truyền qua màng đối với kali, với dòng đi ra của kali và do đó trở lại TRP đưa đến mức độ âm tính nhiều hơn. Mặt khác, kali thấp, làm tăng tốc độ khử cực và có thể gây ra tính tự động trong các sợi tự động tiềm ẩn. Ở phương diện khác, trong thực nghiệm tế bào cơ tim của thỏ, người ta đã nhận thấy khi tăng kali máu đã gây ra sự tăng phân cực.
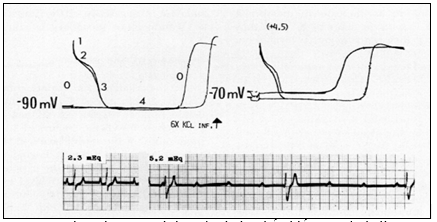
Hình 1 chỉ ra: Hình bên dưới cho thấy blốc AV do kali trong ngộ độc digitalis ở chó. Hình bên trên minh hoạ cơ chế điện sinh lý gây ra blốc. Hai điện thế hoạt động đối chứng được ghi khoảng 1 mm riêng rẽ và phase 0 của chúng được khuếch đại thể hiện ở bên trái. Sau khi truyền kali clorua (bảng bên phải) TRP (giai đoạn 4) đã giảm xuống từ 90 xuống 70 mV; dV/dt của phase 0 đã bị giảm; sự dẫn truyền giữa hai vi điện cực bị chậm trễ nhiều chỉ ra do sự gia tăng khoảng cách giữa hai AP. (Sự xuất hiện không đều của giai đoạn 0 do thực tế AP bị chồng lấn được ghi ở tốc độ thông thường đã bị “xóa”).
Bảng 1
| K+ | Canxi+ | Mg++ | Na+ | |||||
| Parameters | Increased | Decreased | Increased | Decreased | Increased | Decreased | Increased | Decreased |
| lRMP | D | I(V)I)(P) | 0 | 0 | 0 | – | 0 | – |
| dV/dt | D | 1 or O | D | I | D | O | I | D |
| AP amplitude | D | I or 0 | – | – | – | – | I | – |
| AP duratiorn | D | I | D | I | D | I | – | D |
| Rtefractory period | D | I | D | I | – | I | – | – |
| TP | O | I | D | I | – | – | – | – |
| Atornaticity | D | I | I | I | D | – | – | – |
| Cornductivity | ID | D | D | I | D | I | O | D |
| Excitability | ID | ID | D | I | O | O | – | D |
| Important in clinical arrhlythmia | Yes | Yes | May reverse K+ effect ltarely depresses coniduction | – | ? | ? | May revers K+ effect | |
Bảng 1 cho biết ảnh hưởng của điện giải trên các đặc tính điện sinh lý của tim dưới sự thay đổi của các điều kiện thực nghiệm.*
Các chữ viết tắt: RMP: điện thế màng lúc nghỉ; dV/dt: tần số biến đổi phase 0; AP: điện thế hoạt động; TP: điện thế ngưỡng; D: giảm; I: tăng; ID: đầu tiên là tăng, sau đó là giảm; 0: không thay đổi; V: thuộc về thất; P: Purkinje. * Fisch C Greenspan K.[49] Circulation, Volume XLVII, February 1973
Thông thường, các tế bào cơ thất nhạy cảm ít hơn với kali so với cơ nhĩ, các sợi chuyên biệt của nút xoang nhĩ (SA) và bó His nhạy cảm ít nhất. Ngoài ra, có các tế bào trong phạm vi nhĩ, ngoại trừ các tế bào nút SA, chẳng hạn các bó bên trong nút cho thấy trở kháng có chọn lọc với kali và có thể đóng vai trò trong dẫn truyền trong nhĩ và ưu thế trong nút. Sự nhạy cảm của các tổ chức cơ tim đối với kali có thể thay đổi do các yếu tố chẳng hạn như các mức độ của điện giải, pH, độ bão hòa ô xy và tốc độ tăng lên của kali huyết tương. Tất cả cần phải được bao gồm trong bất kỳ sự xem xét sự nhạy cảm tương đối của các tổ chức khác nhau đối với kali.
Tốc độ nhanh chóng với các thay đổi của mức độ kali, không chỉ ở mức độ tuyệt đối, mà quan trọng là dự đoán ảnh hưởng của K lên hình thành xung động và dẫn truyền xung động. Sự gia tăng nhanh chóng của mức độ kali đến bình thường trong truyền bổ xung kali ở người hoặc ở động vật bị giảm kali có thể gây ra nhịp chậm, ngừng tim hoặc ức chế dẫn truyền. Các thay đổi này được bổ xung vào việc tăng lên đột ngột trong sự âm tính của TRP do sự tăng lên của dẫn truyền kali thứ phát do tăng kali ngoại bào.
Tăng kali và dẫn truyền
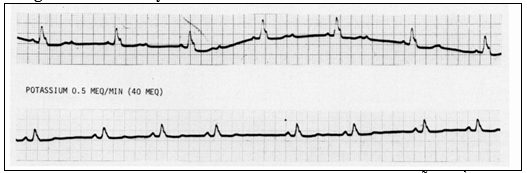
Hình 2 chỉ ra trên ECG ảnh hưởng của tăng nhẹ kali máu lên dẫn truyền A-V. Băng đối chứng chứng tỏ nhịp đều với khoảng thời gian P-P 0,66-0,68 giây và blốc A-V 2:1. Sau khi truyền kali, nhịp xoang với dẫn truyền A –V 1:1 đã được tái lập. Mặc dù khả năng tăng nhẹ kali đã cải thiện dẫn truyền A –V, sau đó đưa đến nhịp xoang chậm hơn (không đều). PP thay đổi từ 1,06 đến 1,50 giây có thể do tác động trực tiếp hoặc ảnh hưởng của kali lên pha 4 ở nút xoang qua trung gian phế vị.
Ở nồng độ 6,0-6,5 mEq/l, tương ứng với tăng kali máu nhẹ, dẫn truyền A-V được gia tăng, cho đến mức khoảng 7,5 imEq/l, dẫn truyền cao hơn bị ức chế. Gia tăng dẫn truyền AV đã được khẳng định trong thực nghiệm của Langendorff trên mẫu tim thỏ, ở thỏ nguyên vẹn và chó đã bị cắt bỏ phế vị bằng tiếp tục truyền ion dương vào tĩnh mạch và truyền trực tiếp vào mạch vành. Dẫn truyền nhanh nhất ở mức kali khoảng 6,0-6,5 mEq/l. Trong tình huống lâm sàng, ảnh hưởng gia tăng của tăng kali trung bình có thể cải thiện dẫn truyền ở các bệnh nhân nhịp xoang và blốc AV (hình 2), hoặc gia tăng dẫn truyền AV và do đó tăng tần số thất ở bệnh nhân rung nhĩ. Vì khó khăn trong việc duy trì phạm vi hẹp trong điều trị kali hiếm gặp, nên từ trước đến giờ, sử dụng để cải thiện dẫn truyền AV ở bệnh nhân blốc AV. Sự gia tăng dẫn truyền có thể giải thích sự tương tác của một số cơ chế, chẳng hạn, hiệu quả chống phế vị của kali và giảm điện thế lúc nghỉ bằng ion dương. Người ta cũng đã nhận thấy kali ức chế ảnh hưởng của acetylcholin ngoại sinh lên dẫn truyền A-V. Tăng kích thích bằng cách đưa TRP gần hơn với điện thế ngưỡng có thể cũng gia tăng tốc độ dẫn truyền.
Ức chế dẫn truyền AV do kali đã được Mathison thông báo lần đầu tiên đầu tiên vào năm 1911, khi đó không cần sự trợ giúp của điện tim, có thể tạo ra dễ dàng trong phòng thí nghiệm. Ở chó, tăng kali huyết tương trên trung bình 8,4 mEq/l, kết quả, gần như đồng nhất, trong sự thay đổi các bất thường dẫn truyên AV. Trong một số trường hợp, blốc AV hoàn toàn được gây ra tại một thời điểm khi cả hai sóng P và QRS đã được ghi lại, cho thấy trong điều kiện của một thiết kế thí nghiệm đặc biệt, hệ thống dẫn truyền AV đề kháng ít với ảnh hưởng ức chế với kali hơn cả tổ chức tâm nhĩ hoặc tâm thất (hình 3).
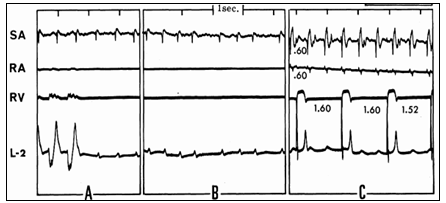
Hình 3 chỉ ra ảnh hưởng khác biệt của truyền kali với tốc độ 1,4 mEq / phút trên các mô tim khác nhau. Các bản ghi thu được từ khu vực xoang nhĩ (SA), tâm nhĩ phải (RA), tâm thất phải (RV) và chuyển đạo II bề mặt (L-2). (A và B) Hoạt động nhĩ và thất bị ức chế ở thời điểm khi điện thế SA và P bề mặt có thể chứng minh rõ ràng. Điều này chỉ ra nút SA và một số phần của tâm nhĩ (trừ các khu vực mà từ đó RA đã được ghi nhận) nhiều khả năng kháng kali hơn các mô thất. (C) Được ghi sau khi ngừng truyền kali, phân ly nhĩ thất hoàn toàn chỉ ra mô bộ nối đề kháng ít hơn với kali so với nút SA, nhĩ hoặc các thất. Hơn nữa, QRS hẹp và sóng P rộng cho thấy tâm thất nhiều khả năng đề kháng kali hơn tâm nhĩ.
Bằng cách ghi lại trực tiếp từ bó His, người ta đã chứng minh nếu kali được truyền với tốc độ khác nhau từ 0,96 đến 1,6 mEq / phút, sự chậm trễ ban đầu và / hoặc blốc dẫn truyền AV ở phía trên bó His, chỉ khi truyền thêm kali tiếp theo mới xuất hiện blốc dưới bó His. Tuy nhiên, nếu truyền kali rất nhanh (2,5 mEq / phút), hầu hết các vị trí bị ức chế dẫn truyền đáng kể có thể được chứng minh đến dưới bó His, ở một thời điểm cho thấy bộ nối bị ức chế ít hơn. Các kết quả tiếp theo gợi ý bất kỳ các tranh luận nào về ảnh hưởng của kali lên các mô khác nhau của tim nên được giải thích do tốc độ truyền.
Các tình huống đồng nhất, trong đó, người ta có thể tạo ra blốc AV ở chó, nhưng lại hiếm được quan sát như vậy trong tăng kali máu lâm sàng. Trong thực tế, đối với sự hiểu biết của chúng ta, trong tăng kali máu lâm sàng “tự phát”, có nghĩa tăng kali máu do quá trình bệnh tự tiến triển chứ không phải do sử dụng kali, blốc gây kéo dài PR đơn thuần nhiều hơn vẫn chưa được ghi nhận. Các trường hợp rải rác blốc AV cao độ đã được chứng minh khi sử dụng kali liều lớn (130-200 mEq) để điều trị các rối loạn nhịp (hình 4) hoặc trong quá trình khảo sát lâm sàng hiệu quả của kali (hình 5). Sự khác biệt này giữa tăng kali máu lâm sàng “tự phát” và thử nghiệm có thể được giải thích do thực tế tăng kali máu thử nghiệm, biểu hiện ảnh hưởng kali “tinh khiết”, trong khi ở những bệnh nhân tăng kali máu thường đi kèm rối loạn cân bằng kiềm toan và nồng độ điện giải khác ngoài kali. Hơn nữa, tần số tăng mức độ kali đóng một vai trò quan trọng. Trong trạng thái thực nghiệm kali huyết tương tăng lên tương đối nhanh, trong khi trong trạng thái bệnh lý kali huyết tương tăng lên rất chậm. Tầm quan trọng của tần số tăng lên của kali huyết tương được ủng hộ bằng quan sát ở một số bệnh nhân có blốc AV độ 2 và độ 3 được tạo ra bằng kali, ion dương được sử dụng nhanh hoặc ở liều lượng lớn lúc nào cũng gây tác động như vậy.
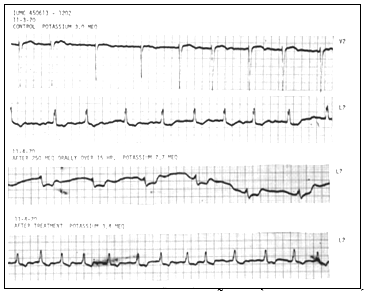
Hình 4 chỉ ra ảnh hưởng kali lên dẫn truyền AV và nội thất ở bệnh nhân bị nhiễm digitalis điển hình. Đường biểu hiện đối chứng (chuyển đạo V2 và L2) chứng tỏ rung nhĩ trong đó đôi khi có nhịp thoát (QRS thứ ba và thứ tư ở chuyển đạo V2). Tiếp theo sử dụng 250 mEq kali qua 15h và với mức độ kali huyết tương 7,7 rnEq / lít, có blốc AV hoàn toàn biểu hiện bằng nhịp thất chậm đều thường xuyên và ức chế dẫn truyền trong thất chỉ ra bằng sự thay đổi trong hình thái và kéo dài QRS. Các thay đổi được phục hồi khi kali huyết tương giảm 3,4; mEq / lít. So với với hình 2, tần số sử dụng kali chậm hơn, nhưng mức độ kali huyết tương đạt đến nhiều khả năng lớn hơn.
Trong thử nghiệm có pha gia tăng dẫn truyền trong thất tạm thời khi kali máu tăng lên vừa phải, nhưng với sự tăng thêm kali dẫn truyền trong thất sẽ bị ức chế. Trong tăng kali máu lâm sàng “tự phát” chỉ có ức chế được quan sát. Thực tế, khi đối mặt với tăng kali huyết tương chậm, cơ chế tử vong là ngừng tim do ức chế dẫn truyền trong thất lan tỏa và đôi khi do rung thất.
Trên ECG bề mặt, ức chế dẫn truyền trong thất biểu hiện bằng kéo dài từ từ, QRS thường giống blốc nhánh bó phải (RBBB) và đôi khi blốc nhánh bó trái (LBBB). Trong so sánh blốc nhánh bỏ không do kali, blốc nội thát do kali máu cao gây ra biểu hiện ức chế lan tỏa và đồng nhất toàn bộ hệ thống dẫn truyền trong thất. Theo đó, toàn bộ QRS, cả hai phần khởi đầu và kết thúc, đều bị kéo dài. Khi ECG trong tăng kali giống với RBBB, phần khởi đầu của QRS sẽ cũng bị kéo dài, trong đó RBBB theo truyền thống không bị ảnh hưởng. Khi ECG giống LBBB, sự xuất hiện của sóng S trên các chuyển đạo thất trái sẽ chỉ ra sự chậm trễ của vị trí kết thúc của QRS, trong khi trong LBBB thông thường chỉ có các phần khởi đầu và giữa bị kéo dài.
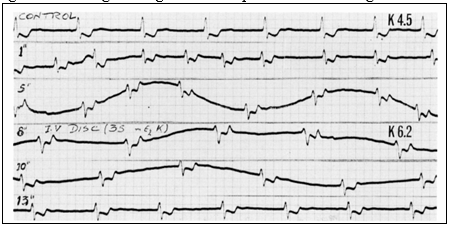
Hình 5 cho thấy ảnh hưởng của truyền kali tĩnh mạch nhanh cho một bệnh nhân bị rung nhĩ. Đường ghi đối chứng (hàng 1, 2) cho thấy rung nhĩ. Sau 5 phút truyền (hàng 4) dẫn truyền thất chậm (QRS bắt đầu mở rộng), tần số thất chậm rõ rệt và có blốc A-V hoàn toàn biểu hiện tần số thất chậm đều trong 10 phút truyền (hàng 6). Ức chế rõ rệt dẫn truyền A-V ở mức kali huyết tương tương đối thấp 6,2 mEq / lít có lẽ là do tốc độ truyền quá nhanh.
Với sự tăng lên đáng kể của kali huyết tương, dẫn truyền trong nhĩ cũng bị ức chế và ECG được đặc trưng bằng kéo dài và giảm biên độ sóng P cũng như kéo dài khoảng PR, cuối cùng biến mất sóng P. Điều này có thể không thấy trên ECG bề mặt để chẩn đoán đặc tính loạn nhịp được tạo ra (hình 6).
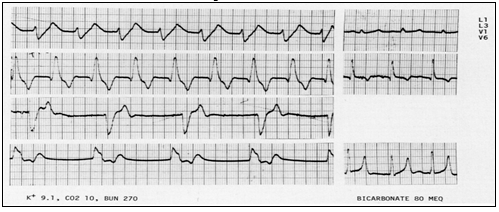
Hình 6 chỉ ra ảnh hưởng của kali huyết tương 9,1 nmEq / lít lên nhịp và dẫn truyền trong thất cũng như sự hồi phục về bình thường bằng truyền 80 mEq natri bicarbonat. Các sóng P vắng mặt. Nguyên nhân kali kéo dài QRS được gợi ý do kéo dài phần khởi đầu và kết thúc của QRS được nhìn rõ nhất ở các chuyển đạo L3 và V6., tương ứng. Người ta không thể chắc chắn về bản chất của rối loạn nhịp. Các khả năng được liệt kê theo thứ tự tần suất: (1) nhịp SA không thể nhận biết được các sóng P trên các chuyển đạo bề mặt với blốc SA 2: 1; ở V1 và V6; (2) nhịp SA với dẫn truyền AV 1: 1 A-V ở Ll và L3 với dẫn truyền 2:1 ở V1 và V6; (3) nhịp xoang chậm ở V1 và V6; hoặc (4) ngưng SA nhĩ ở L1 và L3, và blốc đường ra 2:1 ở V1 và V6.
Truyền Kali ở động vật nguyên vẹn dưới gây mê nhẹ với tầng số tim đối chứng chậm và rối loạn nhịp xoang gây ra nhịp xoang chậm khởi đầu (ở mức độ kali khoảng 6,5 mEq / lít) tiếp theo bằng chậm dần tần số xoang khi kali tăng lên mức 7,5-8,0 mEq / lít. Blốc xoang nhĩ hoặc Wenckebach hoặc Mobitz II giống như được nhìn thấy cả hai trong thực nghiệm và lâm sàng tăng kali “tự phát”. Đây có thể do thực tế các sợi SA có nhiều đề kháng hơn đối với hoạt động ức chế của kali so với các cơ nhĩ. Nói cách khác, xung động SA được tạo ra nhưng bị suy giảm dẫn truyền do ức chế dẫn truyền trong nhĩ. Mặt khác, nút SA đã có nhận cảm nhiều hơn so với cơ nhĩ, ngưng SA có thể là hậu quả. Trong các tình huống lâm sàng, blốc SA do bị động hoặc các nhịp thoát gia tăng có nguồn gốc hoặc ở bộ nối hoặc hệ thống His-Purkinje.
Như với khả năng tăng tốc dẫn truyền, ức chế dẫn truyền có thể là kết quả sự tương tác của một số yếu tố. Các yếu tố này có thể bao gồm, ví dụ, giảm điện thế qua màng lúc nghỉ (TRP) và khả năng hoạt động phế vị.
Hạ kali và dẫn truyền
Ở các động vật thí nghiệm, hạ kali máu có thể gây ra sự chậm trễ và blốc dẫn truyền A-V. Điều này đã được ghi nhận ở lợn cho ăn uống chế độ ít kali, ở tim ếch, loài rùa và thỏ cô lập tiếp xúc với nồng độ kali thấp. Ức chế dẫn truyền trong thất do hạ kali máu ít được thấy rõ rệt nhưng đã được mô tả trên các tim chó, thỏ và rùa. Cơ chế chính xác của chậm dẫn truyền còn chưa rõ ràng, nhưng có thể liên quan đến sự tăng phân cực của tế bào cơ tim. Kết quả, kích thích mạnh hơn cần thiết để đưa TRP đến TP. Đây cũng có thể do sự kích thích của tế bào trước khi khử cực hoàn toàn. Trong tình huống như vậy để đưa điện thế tăng lên được giảm xuống với hậu quả dẫn truyền bị ức chế. Ngược lại, với hạ kali máu thực nghiệm, kéo dài P-R trong bối cảnh lâm sàng là rất hiếm. Một số lượng lớn ECG ở bệnh nhân hạ kali máu đã được nhiều nhà nghiên cứu đánh giá, nhưng kéo dài khoảng P-R có ý nghĩa thống kê không được ghi nhận. Điều này cũng đúng cho cả QRS.
Kali và tính tự động
Sử dụng kali chậm cho động vật gây ra ngừng thất do ức chế dẫn truyền trong thất. Mặt khác, sử dụng nhanh gây ra các ngoại vị có nguồn gốc bộ nối hoặc thất. Sau đó có thể ngừng tim trong rung thất. Tương tự như vậy, sử dụng kali nhanh ở người có thể dẫn đến ngoại vị thất. Các ngoại vị được kèm theo bằng chứng ức chế dẫn truyền do kali tạo ra, gợi ý cơ chế ngoại vị do vào lại hơn là tự động. Nhịp nhanh bộ nối có thể ngoại lệ trong đó, nhịp nhanh này cũng có thể đại diện thực sự cho tăng cường tính tự động. Ngoại vị hiếm gặp trong tăng kali máu lâm sàng tự phát, ngoại trừ biến cố cuối cùng.
Hạ kali máu gây ra nhịp ngoại vị dưới tính đa dạng của các tình huống thực nghiệm và lâm sàng, với một phạm vi lớn các loạn nhịp như xuất hiện loạn nhịp nhĩ, bộ nối và loạn nhịp thất. Các ngoại vị có thể do tính tự động của các sợi tạo nhịp tiềm ẩn gia tăng được cho do dẫn truyền kali bị giảm, với kết quả dòng kali vào bị giảm mặc dù dòng natri vào, đưa đến mất đi sự âm tính bên trong tế bào nhanh nhiều hơn. Ngoài ra, người ta cũng chứng minh thời gian phục hồi của AP có thể vượt quá khoảng thời gian của giai đoạn trơ. Do đó xung được dẫn truyền có thể bị kích động trước khi diễn ra sự phục hồi của AP hoàn toàn, cả vào thời điểm khi TRP bất thường gần sát hơn với TP, do đó, đòi hỏi một yếu tố kích thích “yếu” đối với kích thích. Do hạ kali máu cũng có thể làm giảm dẫn truyền, điều này cũng có thể góp phần vào sự xuất hiện rối loạn nhịp tim bằng vào lại gia tăng.
Ảnh hưởng của kali lên rối loạn nhịp
Sử dụng kali sẽ thường xuyên ức chế loạn nhịp ngoại vị ở động vật. Ức chế này nằm trong phương pháp đo lớn độc lập với mức độ kiểm soát kali và nhiều khả năng độc lập với cơ chế rối loạn nhịp (tự động tính hoặc vào lại). Các tác dụng chống loạn nhịp có phần dự đoán nhiều hơn khi ngoại vị do digitalis. Người ta tin, lý do kali, một chất tương đối đáng tin cậy cho ức chế loạn nhịp do ảnh hưởng của nó lên cả hai tính tự động và dẫn truyền. Trong trường hợp trước đây, kali gây ức chế khử cực pha 4 tự phát bằng cách tăng dẫn truyền kali qua màng. Nếu các rối loạn nhịp tim do vào lại, hoặc gia tăng hoặc ức chế dẫn truyền có thể làm gián đoạn đường vào lại và cắt cơn loạn nhịp. Do đặc tính chống loạn nhịp của kali biểu hiện ở mức 6,0-6,5 mEq / lít, mức độ đó, kali làm gia tăng dẫn truyền, người ta có thể phỏng đoán trong trường hợp loạn nhịp vào lại việc loại bỏ các ngoại vị do cải thiện dẫn truyền ở các nhánh của đường vào lại bị ức chế.
Ở các bệnh nhân có rung nhĩ và hoạt động ngoại vị thất muộn hơn có thể sẽ bị ức chế bằng cơ chế khác, được gọi là sự gia tăng của dẫn truyền AV và tăng tần số thất, tác động tương tự atropine. Ảnh hưởng này có thể rõ do tính chất chống phế vị của các ions dương.
Kali và tính chịu kích thích
Bằng cách làm giảm điện thế lúc nghỉ, kali gây giảm sức mạnh kích thích cần thiết để đưa TRP đến TP và do đó, trong một phạm vi hẹp của tăng kali máu, có thể khôi phục lại sự đáp ứng với kích thích bên ngoài. Điều khó khăn do từ ECG bề mặt để biết sự hồi phục đáp ứng có chắc chắn hay không do tính chịu kích được tăng lên, có thể gây ra suy giảm đáp ứng với kích thích tạo nhịp.
Kali và Digitalis
Các hoạt động của digitalis trên đặc tính điện sinh lý của tim thường xuyên bị ảnh hưởng do sự thay đổi của dự trữ kali. Mối tương quan này có thể trở thành biểu hiện dưới hình thức (1) ức chế ngoại vị do digitalis bằng kali huyết tương tăng lên, (2) ngoại vị gia tăng với nồng độ kali thấp, và (3) tăng khả năng ức chế dẫn truyền do digitalis tạo ra bằng kali huyết tương tăng lên.
Ngay từ năm 1918, Loewi chỉ ra kali có khả năng ức chế nhịp ngoại vị do digitalis gây ra ở các động vật thí nghiệm. Điều này đã được khẳng định trên người trong một loạt các báo cáo của Sampson và cộng sự và sau đó do các tác giả khác. Cơ chế của ức chế loạn nhịp tim do tính tự động có thể liên quan tới sự hoạt hóa của bơm ATPase của kali, với sự gia tăng dòng kali ra ngoài, ức chế pha 4 khử cực và do đó ức chế tính tự động. Trong trường hợp vào lại, có khả năng kali loại bỏ ngoại vị bằng thay đổi dẫn truyền.
Hạ kali máu làm tăng rối loạn nhịp do digitalis gây ra. Độ nhạy được tăng lên này đối với digitalis khi có hạ kali máu đã được chứng minh trong quá trình lọc máu chạy thận nhân tạo, lợi tiểu nhiều, mất kali do một loạt các rối loạn đường ruột và điều chỉnh glucose và steroids. Người ta có thể nhận thấy mất kali nội bào do suy tim (HF) có thể thúc đẩy ngộ độc digitalis, điều này có thể giải thích nhậy cảm của bệnh tim nặng được tăng lên với digitalis.
Các quan sát tăng kali máu, đặc biệt nếu gây ra nhanh chóng, gia tăng ức chế dẫn truyền do digitalis không đáng ngạc nhiên. Cả hai loại thuốc ức chế dẫn truyền và hơn nữa, digitalis bằng cách chẹn kali vận chuyển cho phép tăng kali huyết tương nhanh hơn. Sự gia tăng này đã được nghi nhận về mặt lâm sàng, cả hai với dẫn truyên AV và dẫn truyền trong thất. [2, 27] Ức chế dãn truyền do kali gây ra một cách xuất hiện nổi bật ở động vật ngộ độc digitalis. Sự gia tăng nhanh chóng và rõ ràng của ức chế tạo ra do digitalis do kali được quan sát trong phòng thí nghiệm cần được áp dụng vào các tình huống lâm sàng với các ion dương. Các mức độ khá nghiêm trọng của ngộ độc digitalis và tốc độ nhanh chóng của sự gia tăng của kali huyết tương ít có khả năng gặp trong y học lâm sàng. Bất kể sự khác biệt giữa tình huống thực nghiệm và lâm sàng, sự thật vẫn do kali đưa vào với một tốc độ nhanh không thích hợp cho bệnh nhân ngộ độc digitalis có thể gây ra ức chế dẫn truyền rõ rệt, có thể là xoang nhĩ, trong nhĩ, nhĩ thất hoặc trong thất.
Còn tồn tại phạm vi an toàn có ý nghĩa hơn giữa ngoại vị và các ảnh hưởng ức chế dẫn truyền AV của kali. Ở số lượng lớn các loài động vật được nghiên cứu, các rối loạn nhịp thất được gây ra do acetyl strophanthidin được ức chế bằng kali ở nồng độ trung bình 6,2 mEq/l, trong khi mức độ cần thiết để tạo ra blốc A-V trung bình 8,4 mEq/l. Điều này phù hợp với các ghi nhận sự nhạy cảm khác biệt của các tổ chức Purkinje và bộ nối AV trước đây. Vì vậy, khi đối mặt với rối loạn nhịp nặng do digitalis gây ra, ngay cả trong sự có mặt của dẫn truyền AV chậm trễ đơn thuần, phạm vi an toàn giữa các đặc tính ức chế dẫn truyền và chống ngoại vị của kali cho phép sử dụng kali một cách đúng đắn. (Hình 7).
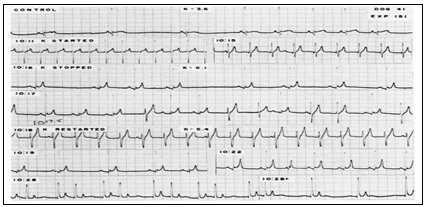
Hình 7 chứng minh loạn nhịp được gây ra do digitalis, ức chế bằng sử dụng đồng nhất với tốc độ 1,2 mEq/rnin, pham vi an toàn giữa hiệu quả ức chế dẫn truyền AV và chống loạn nhịp của kali. Các đường đối chứng đã chứng tỏ loạn nhịp xoang và bắt đầu từ băng 2), tạo ra loạn nhịp ngoại vị bằng acetyl strophanthidin. Sau 5 phút truyền kali và nồng độ kali huyết tương 6,1 mEq/l, loạn nhịp đã bị ức chế, nhưng sau đó trở lại nhanh chóng với việc ngưng truyền kali và kali giảm đến mức 5,4 mnEq/l. Sau 9 phút truyền kali mức 8,2 rnEq/l đã có bằng chứng blốc A-V (hàng dưới).
CANXI
Canxi có tác dụng lên TP, tăng canxi máu làm giảm (âm tính ít hơn) và giảm canxi máu làm tăng lên (âm nhiều hơn) độ lớn của điện thế ngưỡng, nhưng không ảnh hưởng đến hoặc TRP, hình dạng, hoặc biên độ của AP. Điều này thực sự với nồng độ canxi thay đổi từ 1,2 đến 20,8 mEq. Chỉ ở các mức độ nhiều khả năng nhất không phù hợp với sự sống hạ canxi máu gây giảm TRP và tăng độ dốc của khử cực pha 4. Như vậy, trong biến đổi lâm sàng được tính toán ở nồng độ canxi, các ion không có bắt kỳ hiệu quả có ý nghĩa nào lên hoặc biên độ của TRP hoặc khử cực pha 4, hai thay đổi quan trọng trong nguồn gốc rối loạn nhịp.
Ở các động vật thí nghiệm còn nguyên vẹn, tăng canxi máu lên đáng kể có thể gây ra ức chế dẫn truyền trong thất, các co bóp thất sớm, đến rung thất: ở những bệnh nhân với sự tăng lên quá mức của canxi, kéo dài P-R, hoặc blốc AV cao độ và QRS giãn rộng đã được thông báo (hình 8). Tuy nhiên, theo quy luật, tỷ lệ mắc chứng loạn nhịp không tăng lên trong rối loạn chuyển hóa canxi.
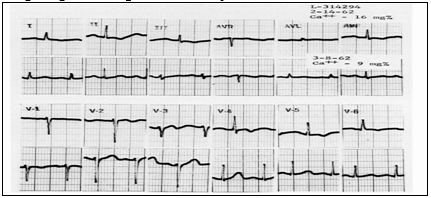
Hình 8 cho thấy khoảng thời gian P-R trong giới hạn bình thường, nhưng kéo dài hơn ở mức độ canxi 16 mg% so với khi mức canxi giảm xuống đến 9 mg%. QRS xuất hiện giãn rộng tối thiểu. Các thay đổi sóng T không đặc hiệu giống như sự thay đổi trong thiếu máu cục bộ cũng được nhìn thấy trong tăng canxi máu. Tất cả những thay đổi được trở lại khi nồng độ canxi về bình thường.
Các gợi ý tăng Canxi làm gia tăng ngoại vị do digitalis cần phải khẳng định. Thực tế làm hạ canxi bằng EDTA có thể đảo ngược loạn nhịp đã ủng hộ như mối liên quan nhưng người ta chưa thể kết luận, khi sự can thiệp này ảnh hưởng đến cả hai loạn nhịp do digitalis và những người không liên quan đến digitalis.
Bằng cách giảm canxi đến 1/20 nồng độ bình thường của nó, một tình huống chưa từng thấy trong y học lâm sàng, rối loạn nhịp có thể được gây ra ở động vật. Một mối quan hệ thú vị và có khả năng hữu ích về mặt lâm sàng tồn tại giữa kali và canxi. Ức chế dẫn truyền và ngoại vị gây ra do kali có thể được phục hồi bằng sử dụng canxi. Tác dụng này của canxi, khi có tăng kali máu, có lẽ liên quan đến sự gia tăng của TRP do canxi.
NATRI
Thay đổi nồng độ natri ảnh hưởng đến biên độ và tần số tăng lên của AP, được tạo nên không có bất kỳ ảnh hưởng đáng kể lên TRP hoặc khử cực pha 4 của tế bào tạo nhịp. Nồng độ natri tăng lên làm tăng biên độ của AP, cũng như tần số của pha 0, trong khi tác dụng ngược lại được ghi nhận với giảm nồng độ natri. Những thay đổi natri gây ra ít có ý nghĩa lâm sàng do mức độ cần thiết để thay đổi điện thế hoạt động thường không phù hợp với sự sống. Do đó, loạn nhịp tim liên quan đến tăng hoặc giam natri máu không được nhận thấy trong y học lâm sàng.
Các mối tương quan của natri và kali có một số hữu ích trong điều trị. Các ức chế dẫn truyền được tạo nên do kali có thể được phục hồi bằng sử dụng natri và nặng lên do nồng độ natri thấp. Trong thực tế lâm sàng, điều này được thể hiện thường xuyên nhất khi sử dụng natri làm hẹp lại QRS, khi nó bị giãn ra do kali. Cơ chế này có thể liên quan đến sự phục hồi của dẫn truyền natri bí suy yếu bằng sử dụng natri với sự gia tăng hợp lực trong tốc độ đột biến của pha 0 và do đó gia tăng dẫn truyền.
MAGIÊ
Tăng magiê đến 3-5 mM/l gây ức chế dẫn truyền A-V. Điều này có thể do chậm tốc độ đột biến của pha 0, cơ chế tương tự hoặc giống hệt với những vấn đề được tạo ra do tăng kali. Có hay không sự thay đổi của nồng độ magiê trong phạm vi lâm sàng gây ra loạn nhịp tim vẫn còn được xem xét. Tuy nhiên, đến nay, có rất ít bằng chứng cho thấy giảm magiê “tự phát” đưa đến loạn nhịp. Nghiên cứu trẻ em bị suy dinh dưỡng, năng lượng – protein và ức chế nồng độ magiê không xuất hiện bất cứ loạn nhịp trừ nhịp xoang nhanh và điều này đã không bị ảnh hưởng do magiê thấp. Mối quan hệ magiê thấp với rối loạn nhịp do digitalis gây ra cần quan tâm. Trong một nghiên cứu, hạ thấp nồng độ magiê bằng lọc máu với magiê hòa tan tự do đã làm giảm số lượng acetyl strophanthidin cần thiết để gây rối loạn nhịp tim. Rối loạn nhịp tim đã nhanh chóng hủy bỏ ở phần lớn, nhưng không phải tất cả ở các loài động vật bằng cách truyền magiê sulfate. Giảm magiê là nghiêm trọng và xảy ra nhanh chóng và liệu những kết quả có thể được suy ra cho các tình huống lâm sàng vẫn cần được nhìn nhận xem xét. Các câu hỏi về các đặc trưng của magiê cho loạn nhịp digitalis cũng cần làm sáng tỏ hơn nữa, vì như trong trường hợp của kali, magiê ức chế loạn nhịp do digitalis, cũng như những người không phải do các glycoside.
Sự ra đời của điện tâm đồ, vi điện cực và kỹ thuật cho các nghiên cứu về các dòng ions chuyển qua màng đã góp phần vào sự hiểu biết của chúng ta về một số trong những mối quan hệ phức tạp giữa điện giải đồ thay đổi và rối loạn nhịp. Mô phỏng các trạng thái lâm sàng trong đó rối loạn nhịp được ghi lại (ví dụ rối loạn đồng thời một số chất điện giải với các biến số như pH, độ bão hòa ô xy, chất thải trao đổi chất, tình trạng tổ chức bệnh của mô) ở cả động vật còn nguyên vẹn và mô bị cô lập chúng ta sẽ hiểu thêm về cơ chế điện sinh lý của loạn nhịp. Đây chỉ là một trong nhiều con đường mở ra cho chúng ta để nghiên cứu về căn nguyên, duy trì và kiểm soát các rối loạn nhịp.
CÁC NGUYÊN NHÂN GÂY HẠ KALI MÁU Ở NGƯỜI LỚN
Giảm kali máu là một vấn đề lâm sàng thông thường. Kali vào cơ thể theo đường ăn uống hoặc truyền tĩnh mạch, phần lớn được lưu trữ trong tế bào, sau đó thải tiết ra nước tiểu. Vì vậy, cung cấp theo đường ăn uống giảm, tăng vẫn chuyển vào tế bào, hoặc thường nhất, tăng mất đi theo đường tiểu, ống tiêu hóa, hoặc theo mô hôi có thể dẫn đến giảm nồng độ kali huyết tương. Các nguyên nhân được thống kê ở bảng 2.
Bảng 2.
| Giảm cung cấp kali |
| Kali đi vào trong tế bào nhiều |
| Tăng pH ngoại bào |
| Tính khả dụng insulin tăng lên |
| Hoạt động β-adrenergic tăng lên – stress hoặc sử dụng ức chế béta |
| Liệt chu kỳ do hạ kali máu (Hypokalemic periodic paralysis) |
| Sản xuất tế bào máu tăng lên đáng kể |
| Hạ thân nhiệt |
| Nhiễm độc Chloroquine |
| Mất qua đường tiêu hóa tăng lên |
| Nôn ói |
| Tiêu chẩy |
| Qua các ống dẫn lưu |
| Lạm dụng thuốc nhuận trường |
| Mất qua đường tiểu tăng lên |
| Lợi tiểu |
| Cường mineralocorticoid tiên phát |
| Mất chế tiết dạ dầy |
| Không tái hấp thu các anions |
| Toan hóa ông thận |
| Giảm magiê |
| Amphotericin B |
| Các bệnh thận mất muối như: – hội chứng Bartter’s hoặc Gitelman’s |
| Đa niệu (Polyuria) |
| Mất theo mồ hôi tăng lên |
| Các trạng thái thẩm tách (Dialysis) |
| Tách hồng cầu khỏi huyết tương (Plasmapheresis) |
Giảm do ăn uống
Cung cấp kali theo ăn uống hàng ngày bình thường từ40 đến 120 mEq, phần lớn sau đó được thải qua đường tiểu. Thận có thể giảm thải kali tối thiểu là 5-25 mEq mỗi ngày khi có giảm kali máu. Do đó, giảm cung cấp đơn thuần hiếm khi gây hạ kali máu đáng kể. Điều này đã được chứng minh trong nghiên cứu ở các cá nhân bình thường, ở người giảm cung cấp kali đến 20 mEq mỗi ngày có liên quan với giảm kali huyết thanh từ 4,1 mEq/l ở mức cơ bản lên 3,5 mEq/l. Tuy nhiên cung cấp kali giảm có thể thúc đẩy làm nặng lên tình trạng thiếu hút kali khi có các nguyên nhân khác như dùng lợi tiểu.
Tăng cường đi vào tế bào — Hơn 98% tổng số kali cơ thể ở trong tế bào, chủ yếu trong cơ bắp. Sự phân bố bình thường của kali giữa các tế bào và dịch ngoại bào chủ yếu được duy trì bằng bơm Na-K-ATPase ở màng tế bào. Tăng hoạt động của bơm Na-K-ATPase và / hoặc thay đổi trong các đường vận chuyển kali khác có thể dẫn đến hạ kali máu thoáng qua do tăng nhập kali vào trong tế bào.
Tăng tính khả dụng của insulin —Insulin thúc đẩy sự xâm nhập của kali vào tế bào cơ xương và gan, chủ yếu bằng cách tăng các hoạt động của bơm Na-K-ATPase. Hiệu ứng này thể hiện rõ nhất sau khi tiêm insulin ngoại sinh cho bệnh nhân tiểu đường nhiễm ceton acid hoặc tăng đường huyết nặng không nhiễm ceton thường có thể thấy kali bình thường hoặc tăng, mặc dù họ đã mất kali. Hạ kali máu hiếm khi có thể gây ra do quá liều insulin.
Insulin nội sinh tiết ra để đáp ứng với chuyển hóa carbohydrate cũng có thể góp phần giảm kali huyết. Điều này đã được mô tả ở những bệnh nhân với hội chứng tái nuôi dưỡng và sau khi truyền tĩnh mạch kali trong dextrose trong dung dịch nước. Vì vậy, liệu pháp truyền kali tĩnh mạch ban đầu cho hạ kali máu nên được sử dụng trong một dung dịch nước muối chứ không phải dung dịch dextrose.
Hoạt động beta-adrenergic tăng lên — Catecholamin nội sinh, hoạt hóa thông qua beta-2 adrenergic, có thể thúc đẩy kali vào tế bào bằng cách tăng các hoạt động của bơm Na-K-ATPase và K-2Cl Na vận chuyển (NKCC1) và có thể bằng cách tăng giải phóng insulin. Vai trò sinh lý được đề xuất cho hiệu ứng hoạt động beta-2 adrenergic được tăng lên này để làm điều tiết tăng kali cấp tính do găng sức.
Khi gây ra lượng kali vào các tế bào, hạ kali máu thoáng qua có thể phát triển ở bất kỳ tình huống nào tiết epinephrine do stress gây ra, gổm cả ngừng uống rượu (alcohol withdrawal), nhồi máu cơ tim cấp tính, chấn thương đầu, hoặc nhiễm độc theophylline.
Tác dụng tương tự, trong đó nồng độ kali huyết thanh có thể suy giảm cấp thời đến hơn 0,5-1 mEq/l, đã được thấy trong các tình huống sau đây:
● Sau khi sử dụng chủ vận beta-adrenergic ngoại sinh (như albuterol, terbutaline, hoặc dobutamine) để điều trị bệnh hen phế quản hoặc suy tim hoặc để ngăn ngừa sinh non.
● Uống các chất giao cảm thông thường được thấy trong thuốc thông mũi hoặc các chất ăn kiêng, như pseudoephedrine và ephedrine.
● Sử dụng heroin hoặc thịt bị nhiễm các chất chủ vận beta, clenbuterol.
Bất kể nguyên nhân của tăng hoạt động beta-adrenergic, mức độ hạ kali máu sẽ lớn hơn ở những bệnh nhân hạ kali từ trước, ví dụ, để điều trị lợi tiểu (hình 9).
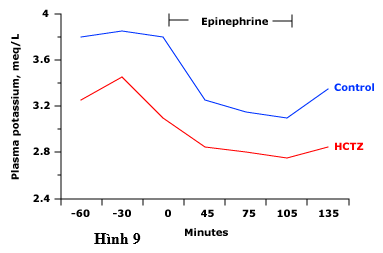
Hình 9 biểu thị nồng độ kali huyết tương trong truyền epinephrine (ở liều sinh lý) ở bệnh nhân điều trị bằng giả dược hoặc thuốc lợi tiểu thiazide trong bảy ngày. Nồng độ kali trong huyết tương giảm ở cả hai nhóm, nhưng khả năng đạt mức độ nguy hiểm ở các bệnh nhân được điều trị thuốc lợi tiểu-người đã có hạ kali máu nhẹ.
Ảnh hưởng gây hạ kali của các thuốc chủ vận beta có khả năng đặc biệt gây rối loạn nhịp trong các tình huống sau:
● Khi dùng đồng thời với thuốc lợi tiểu ở bệnh nhân tăng huyết áp. Thường gặp ở các bệnh nhân điều trị tăng huyết áp có COPD
● Ở sản phụ sinh non. Một nghiên cứu, ví dụ, cho thấy 3 trong số 14 bệnh nhân điều trị bằng hoặc ritodrine hoặc terbutaline để phòng ngừa sinh non phát triển rối loạn nhịp tim có triệu chứng, chẳng hạn như nhịp tim nhanh trên thất hoặc các phức hợp nhĩ hoặc thất sớm.
Ngoài ra, chất chủ vận beta có thể được điều trị cấp thời các tăng kali nặng. Mặt khác, thuốc chẹn beta làm tăng kali huyết thanh, đặc biệt nếu được dùng thêm kali (hình 10).
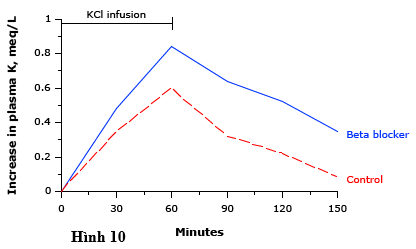
Hình 10 biểu thi thay đổi trong nồng độ kali trong huyết tương sau khi truyền kali clorua ở nhóm chứng (đường không liên tục) và những người điều trị với thuốc chẹn beta (đường liền). Sự gia tăng nồng độ kali huyết thanh lớn hơn một cách có ý nghĩa khi có mặt của các chẹn beta.
pH ngoại bào tăng lên —Hoặc nhiễm kiềm chuyển hóa hoặc hô hấp có thể thúc đẩy kali vào trong tế bào. Khi có nồng độ ions hydro giảm xuống, các ion hydro ra khỏi tế bào để giảm thiểu sự gia tăng pH nội bào; sự cần thiết phải duy trì thần kinh điện học sau đó đòi hỏi sự xâm nhập của một số kali (và natri) vào tế bào. Nồng độ kali huyết thanh giảm ít hơn 0,4 mEq/L cho mỗi tăng 0,1 đơn vị pH.
Ngoài giảm ions hydro dẫn đến sự hấp thu kali của tế bào, nhiễm kiềm chuyển hóa thường thấy ở hạ kali máu, chủ yếu do những nguyên nhân chính của hạ kali máu (như thuốc lợi tiểu, nôn và cường aldosteron, được mô tả dưới đây) cũng dẫn đến suy giảm ion hydro. Hạ kali máu là một đồng yếu tố quan trọng trong việc duy trì nhiễm kiềm chuyển hóa vì nó thúc đẩy sự tái hấp thu bicarbonate, do đó làm suy yếu sự điều chỉnh hạ ions hydro do bài tiết của bicarbonate dư thừa (hình 11).
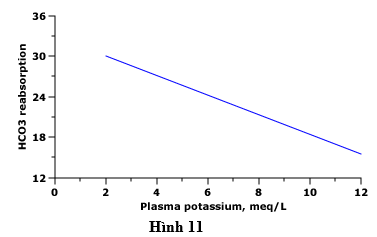
Hình 11 biểu thị mối quan hệ nghịch đảo giữa kali huyết tương và tái hấp thu bicarbonate. Tái hấp thu HCO3 ở ống thận (mEq/l của dịch lọc cầu thận) như là một chức năng của nồng độ kali trong huyết tương.
Liệt cơ chu ký do hạ kali máu (Hypokalemic periodic paralysis) — Liệt chu kỳ do hạ kali-máu là một rối loạn thần kinh cơ hiếm, đặc trưng bằng các cơn có khả năng gây tử vong do yếu cơ hoặc liệt có thể ảnh hưởng đến các cơ hô hấp. Bệnh có thể di truyền, với nhiễm sắc thể thường kiểu autosomal trội, hoặc mắc phải ở các bệnh nhân cường giáp. các cơn cấp tính, trong đó các chuyển động đột ngột của kali vào trong tế bào có thể làm giảm kali huyết thanh thấp đến 1,5-2,5 mEq/l, thường bị thúc đẩy sau gắng sức, căng thẳng, hoặc một bữa ăn carbohydrate. Kali huyết thanh bình thường giữa các cơn liệt, một đặc điểm có thể giúp phân biệt liệt chu kỳ từ các hình thức khác của liệt hạ kali-máu. Bệnh nhân mắc bệnh tê liệt chu kỳ hạ kali-máu cũng sẽ có bài tiết kali nước tiểu thấp. Phát hiện này có thể giúp phân biệt các bệnh nhân từ những người có liệt hạ kali-máu do mất kali theo đường thận.
Sản xuất tế bào máu tăng — Sự gia tăng cấp tính trong sản xuất tế bào máu có liên quan đến sự hấp thu kali do các tế bào mới và điều này có thể dẫn đến hạ kali máu. Điều trị vitamin B12 hoặc acid folic để điều trị thiếu máu cầu khổng lồ hoặc sử dụng các bạch cầu hạt và đại thực bào yếu tố kích thích xâm thực (GM-CSF) để điều trị giảm bạch cầu gây ra các tình trạng xảy ra phổ biến nhất.
Cơ chế tương tự có thể dẫn đến hạ kali máu giả ở các mẫu máu lấy từ bệnh nhân với nhiều tế bào máu hoạt động trao đổi chất, chẳng hạn như bệnh nhân bị bệnh bạch cầu tủy cấp tính và một số lượng tế bào bạch cầu cao. Nếu máu được phép giữ ở nhiệt độ phòng trước khi đo nồng độ kali, giá trị đo có thể dưới 1 mEq/l mặc dù bệnh nhân không có triệu chứng, dấu hiệu, hoặc di chứng của hạ kali máu. Hạ kali máu giả này có thể được ngăn ngừa bằng cách hoặc là tách nhanh chóng huyết tương từ các tế bào sau tĩnh mạch, hoặc lưu trữ máu ở 4°C trước khi xét nghiệm.
Hạ thân nhiệt — Tình cờ hoặc gây hạ thân nhiệt có thể đưa kali vào trong tế bào và làm giảm nồng độ kali huyết thanh đến 3,0-3,5 mEq/l hoặc thấp hơn. Ngược lại, bệnh nhân hạ thân nhiệt cơ bản do tử vong và do đó không đáp ứng với hồi sức có thể đã có tăng kali máu trước khi tử vong, do hoại tử mô không thể đảo ngược.
Nhiễm độc bari (barium) — Nhiễm độc, thường do kết quả từ việc ăn thức ăn bị ô nhiễm hoặc do cố ý dùng, có thể gây hạ kali máu bằng cách ngăn chặn các kênh kali trong màng tế bào thường cho phép kali tế bào khuếch tán vào dịch ngoại bào. Điều trị ngộ độc bari với sự thay thế kali phục vụ cho cả hai làm tăng nồng độ kali huyết thanh và chuyển rời bari từ các kênh kali bị ảnh hưởng. Chạy thận nhân tạo, một điều trị hiệu quả.
Bệnh nhân trải qua các thủ tục chụp XQ không có nguy cơ biến chứng này kể từ khi sử dụng sulfat bari trong các thủ thuật này không đi vào hệ tuần hoàn.
Nhiễm độc cesium —Nhiễm độc cesium, thường là do việc sử dụng các cesium chloride như một điều trị thay thế cho bệnh ung thư, có thể được kết hợp với hạ kali máu. Điều này xẩy ra như thế nào còn không rõ, nhưng có thể kết quả của phong tỏa của cesium các kênh kali màng.
Nhiễm độc chloroquine — Hạ kali máu, với nồng độ kali huyết thanh giảm xuống dưới 2,0 mEq/l trong trường hợp nghiêm trọng, một phát hiện phổ biến trong nhiễm độc chloroquine cấp. Ảnh hưởng này này có lẽ qua trung gian bằng chuyển rời kali vào tế bào và có thể bị trầm trọng hơn do sử dụng epinephrine để giúp điều trị nhiễm độc.
Các thuốc chống loạn thần (Antipsychotic drugs) — Hạ kali máu một biến chứng hiếm gặp của điều trị với các thuốc chống loạn thần được lựa chọn, chẳng hạn như risperidone và quetiapine.
MẤT THEO ĐƯỜNG TIÊU HÓA TĂNG LÊN –Mất dịch tiết dạ dày hoặc ruột từ bất kỳ nguyên nhân (nôn mửa, tiêu chảy, thuốc nhuận tràng, hoặc ống dẫn lưu) có liên quan với mất kali và có thể hạ kali máu.
Mất do đường tiêu hóa trên — Nồng độ kali trong dịch tiết dạ dày chỉ 5-10 mEq/l; do đó, sự suy giảm kali trong môi trường này chủ yếu mất theo đường niệu được tăng lên.
Các trình tự tiếp theo chịu trách nhiệm cho mất mát kali niệu với việc di dời acid dạ dày. Các nhiễm kiềm chuyển hóa có liên quan và tăng nồng độ bicarbonate huyết tương tăng tái lọc bicarbonate trên ngưỡng tái hấp thu của nó. Kết quả là, nhiều natri bicarbonate và nước được cung cấp cho các vị trí tiết kali ở đầu xa. Trong sự kết hợp với một sự gia tăng gây ra do giảm thể tích trong tiết aldosterone, hiệu quả toàn bộ làm tiết kali tăng lên và mất kali đường niệu có khả năng lớn. Do sự tiêu hao natri không phù hợp với bicarbonate, nồng độ clorua niệu thấp được sử dụng để phát hiện sự hiện diện của sự suy giảm thể tích.
Tiêu hao kali niệu được nhìn nhận với sự mất của dịch tiết dạ dày thường nổi bật nhất trong những ngày đầu tiên; sau đó, dự trữ tái hấp thu bicarbonate tăng lên, dẫn đến giảm đáng kể trong mất natri, bicarbonate và kali niệu. Tại thời điểm này, độ pH nước tiểu suy giảm từ trên 7,0 (do tiêu hao bicarbonate) xuống dưới 6,0.
Mất theo đường tiêu hóa dưới — Ngược lại với dịch tiết dạ dày, nồng độ kali trong ruột bị mất đi tương đối cao (20-50 mEq/l) trong hầu hết các trường hợp. Ngoài ra, hạ kali máu do mất đi ở ống dạ dầy ruột thấp hơn (thường do tiêu chẩy) thường được kết hợp với tiêu hao bicarbonate và toan chuyển hóa tăng clo máu hơn là nhiễm kiềm chuyển hóa được quan sát với sự mất đi ở dạ dẩy ruột cao hơn. Tuy nhiên, một số bệnh nhân bị hạ kali máu do tiêu chảy tự gây ra hoặc lạm dụng thuốc nhuận tràng âm thầm phát triển nhiễm kiềm chuyển hóa thông qua một cơ chế còn chưa chắc chắn.
Hạ kali máu do mất ở ống tiêu hóa thấp hơn là phổ biến nhất khi mất xảy ra trong một thời gian dài, như với adenoma villous, một peptide ruột vận mạch do khối u tiết ra (VIPoma), hoặc tiêu chảy nhiễm trùng dai dẳng. Trong nhiều trường hợp, mất theo phân được tăng lên không thể giải thích toàn bộ thâm hụt kali. Vì hầu hết những người ăn trung bình 80 mEq kali mỗi ngày và kali bài tiết nước tiểu có thể giảm xuống dưới 15-25 mEq/ngày khi có hạ kali máu, mất theo phân (thường khoảng 10 mEq/ngày) có thể vượt quá 55 65 mEq/ngày để trực tiếp gây hạ kali máu. Tuy nhiên, nhiều bệnh nhân bị tiêu chảy, người trở thành hạ kali-máu có bài tiết kali phân dưới mức này, chỉ ra các yếu tố khác, chẳng hạn như tiêu thụ giảm và giảm thể tích với tăng hoạt động aldosterone, cũng đóng một vai trò cấu thành.
Các nguyên nhân hạ kali máu do mất ở ống tiêu hóa dạ dầy ruột ở thấp hơn được kể đến bao gồm:
● Làm sạch ruột để nội soi bằng cả hai sodium phosphate (bây giờ ít sử dụng) và polyethylene glycol (PEG) để chuẩn bị, đặc biệt là ở người già.
● Viêm đại tràng giả tắc nghẽn (hội chứng Ogilvie), được kết hợp với tiêu chảy do tiết đặc trưng bằng hàm lượng kali cao bất thường do hoạt hóa bài tiết kali ở ruột.
● Ăn đất sét (“geophagia”), đất sẽ liên kết với kali trong đường tiêu hóa.
MẤT DO ĐƯỜNG NIỆU TĂNG LÊN — Nước tiểu bài tiết kali có nguồn gốc chủ yếu từ tiết kali ở ống thận xa, đặc biệt các tế bào chính ở chỗ nối các ống góp và các ống lượn vùng vỏ. Hạ kali máu do mất ở đường niệu chủ yếu là do hai yếu tố:
● Tăng hoạt động mineralocorticoid – Aldosterone hoạt động chủ yếu bằng cách kích thích tái hấp thu natri qua kênh natri biểu mô (ENAC). Tái hấp thu natri làm cho lòng điện âm tương đối, qua đó thúc đẩy bài tiết kali thụ động từ các tế bào hình ống vào trong lòng thông qua các kênh kali (ví dụ, ROMK) trong màng lòng ống (hình 12).
● Cung cấp natri và nước ở đầu xa tăng lên – Tăng cường bài tiết kali có thể dẫn đến việc gia tăng cung cấp natri đến ống góp, đặc biệt nếu đi kèm với các ions âm clorua không được tái hấp thu hơn.
Tiêu hao kali cũng có thể do đa niệu như chúng ta đã biết, ví dụ, trong uống nhiều tiên phát. Trong bối cảnh này, nồng độ kali nước tiểu thấp phù hợp (dưới 15 mEq / L), nhưng bài tiết kali cộng lại có thể lớn hơn lượng đưa vào cơ thể do sự gia tăng đáng kể thể tích nước tiểu.
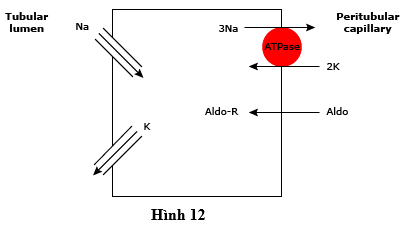
Hình 12 sơ đồ biểu diễn vận chuyển natri và kali vận trong ở các tế bào tái hấp thu natri chính ở các ống góp. Sự tham gia của natri được lọc vào các tế bào này qua trung gian các kênh natri chọn lọc trong màng đỉnh (ENAC); năng lượng cho quá trình này được cung cấp bằng độ chênh (gradient) điện hóa ưu thế cho natri (tế bào bên trong điện âm tính và nồng độ natri tế bào thấp). Tái hấp thu Na được bơm ra khỏi tế bào bằng bơm Na-K-ATPase trong màng (quanh các ống) nền các thành. Tái hấp thu ions dương Na làm cho điện âm trong lòng ống , do đó tạo ra sự chênh lệch (gradient) thuận lợi cho sự bài tiết K vào trong lòng thông qua các kênh kali (ROMK và BK) trong màng đỉnh. Aldosterone (Aldo), sau khi kết hợp với các thụ thể mineralocorticoid cytosolic (Aldo-R), dẫn đến tăng cường tái hấp thu natri và tiết kali bằng cách tăng cả về số lượng các kênh natri mở và số lượng bơm Na-K-ATPase. Các thuốc lợi tiểu tiết kiệm kali (amiloride và triamterene) hoạt động bằng cách trực tiếp ức chế kênh natri biểu mô; spironolactone hoạt động bằng cách cạnh tranh với aldosteron để liên kết với các thụ thể mineralocorticoid.
Ở đại đa số bệnh nhân, ở các hai hoạt động corticoid muối và dòng ra thể hiện tăng lên đồng thời. Nếu chỉ có một tham số được tăng lên, còn lại phải được bình thường gây mất quá nhiều kali. khái niệm quan trọng này được minh họa bằng sự vắng mặt của hạ kali máu ở những bệnh nhân có tăng bù trong aldosterone do giảm thể tích. Trong trường hợp như vậy, angiotensin II kích thích tái hấp thu natri ở ống lượn gần và xa, do đó gián tiếp làm giảm natri và phân phối nước ở xa. Angiotensin II cũng ức chế kênh tiết kali ở các ống góp, bằng cách đó tái cân bằng ảnh hưởng kích kích của aldosterone trên sự tiết kali.
Để đối phó với hạ kali máu do đưa đến bị tăng lên nhưng mất qua đường niệu ổn đinh, tiết kali đường niệu cuối cùng cũng suy sụp đưa đến giá trị tương đương với lượng kali cung cấp vào. Tại thời điểm này, kali huyết thanh thấp nhưng sẽ không suy giảm thêm, trừ khi lượng tiêu hao kali tăng, ví dụ, do tăng liều thuốc lợi tiểu hoặc trong tiết aldosterone ở bệnh tăng aldosterone tiên phát.
Phản ứng thích nghi đối với hậu quả hạ kali này một phần do giảm trong hoạt động của các kênh tiết kali (ví dụ, ROMK) trong màng phía trong lòng của các ống góp phần vỏ thận (hình 12), một hiệu ứng có khả năng qua trung gian do hoạt động angiotensin II được tăng lên. Như một ví dụ, ức chế tuyến yên của các kênh kali lòng ống sẽ làm tối thiểu mức độ hạ kali máu được thấy rõ ở các bệnh nhân bị cường aldosterone tiên phát và có thể giải thích lý do tại sao hạ kali máu là một phát hiện không phù hợp trong rối loạn này.
Các nguyên nhân lớn hao tổn kali đường niệu — Các nguyên nhân phổ biến nhất làm hạ kali máu do mất kali niệu gồm sử dụng thuốc lợi tiểu, tăng hoạt động các corticoid muối tiên phát, tăng cung cấp anion không được tái hấp thu và mất các chất tiết của dạ dày.
Các thuốc lợi tiểu — Bất kỳ thuốc lợi tiểu nào có tác động gần đến vị trí tiết kali – các chất ức chế carbonic anhydrase (ví dụ, acetazolamide), thuốc lợi tiểu quai và các loại lợi tiểu thiazide, cả hai sẽ tăng cung ứng đầu xa và qua việc tạo ra suy giảm thể tích, kích hoạt hệ thống renin-angiotensin-aldosterone. Kết quả, bài tiết kali niệu sẽ tăng lên, dẫn đến hạ kali máu nếu mất theo đường niệu lớn hơn lượng cung cấp vào.
Tỷ lệ và mức độ nghiêm trọng của hạ kali máu phụ thuộc liều, xảy ra ít thường xuyên hơn với liều thấp hơn. Như vậy, liều thiazide thấp hơn (ví dụ, 12,5-25 mg/ngày chlorthalidone hoặc hydrochlorothiazide) hiện đang sử dụng rộng rãi trong điều trị tăng huyết áp, vì liều này hiệu quả trong việc giảm huyết áp (hình 13) với một ảnh hưởng nhỏ hơn vào cân bằng điện giải. Trong hai nghiên cứu tăng huyết áp lớn. chlorthalidone liều thấp, hạ kali máu đòi hỏi phải điều trị xảy ra trong 7-8% bệnh nhân.
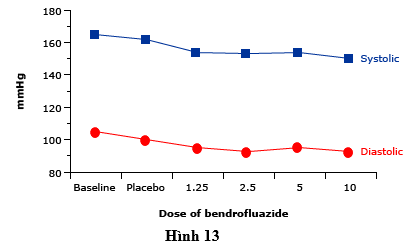
Hình 13 biểu thịđáp ứng hạ huyết áp đối với bendrofluazide liên quan đến liều dùng hàng ngày (mg, nhân với 10 để có được liều lượng tương đương gần đúng của hydrochlorothiazide). Liều ban đầu là 1,25 mg/ngày làm giảm huyết áp so với giả dược; Tuy nhiên, liều lượng cao gây ra đáp ứng chống huyết áp tiếp theo ít hơn. Mỗi nhóm điều trị chứa khoảng 52 bệnh nhân.
Ở những bệnh nhân ổn định ở một liều thuốc lợi tiểu cố định, mất kali, giống như biến chứng điện giải và dịch tạo ra do lợi tiểu khác, chỉ xuất hiện trong 2 tuần lễ đầu tiên điều trị trước khi một trạng thái ổn định mới được thiết lập. Do đó, một bệnh nhân ổn định với nồng độ kali huyết thanh bình thường tại ba tuần không phải là nguy cơ hạ kali máu trễ trừ khi tăng liều thuốc lợi tiểu, mất kali ngoài thận gia tăng, hoặc lượng kali chế độ ăn uống giảm.
Khi trạng thái đã đạt được ổn định, không cần thiết theo dõi thêm, trừ khi tăng liều thuốc lợi tiểu, mất kali ngoài thận tăng lên, hoặc lượng kali chế độ ăn uống giảm. Ví dụ, mất tăng lên và lượng ăn uống giảm xuống trong viêm dạ dày ruột. Ở những bệnh nhân như vậy, tạm ngừng điều trị lợi tiểu một vài ngày có thể thích hợp.
Ở những bệnh nhân tăng huyết áp phát triển hạ kali máu do thuốc lợi tiểu gây ra, hoặc sử dụng các thuốc khác, hoặc điều trị bằng thuốc lợi tiểu tiết kiệm kali hoặc bổ sung kali.
Hoạt động của các hormone khoáng (mineralocorticoid) tăng lên —Hao phí kali theo đường niệu đặc trưng của bất ký trạng thái nào kết hợp với tăng tiết các hormome khoáng chất tiên phát, như với u vỏ thượng thận (adrenal adenoma) gây tăng tiết aldosterone. Các bệnh nhân này thường tăng huyết áp, chẩn đoán phân biệt gồm điều trị lợi tiểu (có thể âm thầm) ở bệnh nhân có bệnh tăng huyết áp cơ bản, bệnh mạch máu thận, trong đó tăng tiết renin dẫn đến tăng cường tiết aldosterone.
Các ions âm không tái hấp thu (Nonreabsorbable anions) — Độ chênh (gradient) điện học âm tính trong lòng ống đã bạo ra không tái hấp thu natri ở ống góp ở vỏ đã bị suy yếu một phần do không tái hấp thu clorua. Tuy nhiên, có một số trạng thái lâm sàng trong đó natri được biểu hiện đến các cầu thận với số lượng tương đối lớn các ions âm không tái hấp thu, gồm bicarbonate với nôn mửa hoặc toan ống lượn đầu gần, beta-hydroxybutyrate trong nhiễm toan ceton do tiểu đường, hippurate sau sử dụng toluene, hoặc một dẫn xuất penicilin ở bệnh nhân được điều trị với penicillin liều cao. Ở những trạng thái này, nhiều natri được cung cấp sẽ được hấp thụ lại để trao đổi với kali, dẫn đến một sự gia tăng khả năng đáng kể trong việc thải kali. Như ví dụ, nồng độ kali huyết thanh dưới 2 mEq/l trong gần một phần tư số bệnh nhân bị nhiễm toan chuyển hóa do toluene gây ra..
Ảnh hưởng của các anion không được tái hấp thu có khả năng chiếm ưu thế nhất khi có suy giảm thể tích đồng thời. Trong bối cảnh này, việc giảm cung cấp clorua ở đầu xa (qua đó hạn chế khả năng tái hấp thu clorua để tiêu tan chênh lệch (gradient) âm tính trong lòng ống) và tiết aldosterone gia tăng cả hai đều thúc đẩy tiết kali. Ví dụ, trong tiểu đưởng nhiễm toan ceton, cung cấp natri và nước ở đầu xa gia tăng (do lợi tiểu thẩm thấu glucose), cường aldosteron giảm thể tích tuần hoàn gây ra và hoạt hóa beta-hydroxybutyrate như một anion không tái hấp thu tất cả có thể góp phần tăng tiêu hao kali.
Mất chế tiết dạ dày – Cơ chế mất chế tiết dạ dày dẫn tiêu hao kali đường niệu được mô tả ở trên, trong đó có sự gia tăng trong cung cấp natri bicarbonate đến ống góp. Vấn đề này thường rõ ràng từ bệnh sử. Tuy nhiên, nếu bệnh sử không hữu ích, chẩn đoán phân biệt ở bệnh nhân huyết áp bình thường với hạ kali máu, tiêu hao kali đường niệu và nhiễm kiềm chuyển hóa bao gồm ói mửa kín đáo, cũng như sử dụng thuốc lợi tiểu và hội chứng Bartter.
Các nguyên nhân ít gặp hơn của tiêu hao kali đường niệu — Các nguyên nhân ít gặp hơn của tiêu hao kali do mất qua đường niệu gồm các rối loạn đa dạng mắc phải và di truyền.
Đa niệu (Polyuria) — Người bị cạn kiệt kali có thể giảm một cách bình thường nồng độ kali niệu đến tối thiểu là 5-10 mEq/l. Tuy nhiên, nếu lượng nước tiểu quá 5 đến 10 lít/ngày, sau đó mất kali bắt buộc có thể vượt quá 50-100 mEq mỗi ngày. Vấn đề này rất có thể xảy ra trong uống nhiêu tiên phát (thường từ tâm lý), trong đó lượng nước tiểu có thể tăng trong thời gian dài. Một mức độ tương đương của đa niệu cũng có thể xảy ra trong đái tháo nhạt trung ương, nhưng những bệnh nhân bị rối loạn này thường tìm kiếm sự chăm sóc y tế ngay sau khi đa niệu đã bắt đầu.
Nhiễm toan ống thận – Tiêu hao Kali có thể xảy ra ở cả hai nhiễm toan ống thận xa (type 1) và gần (type 2). Trong mỗi rối loạn này, mức độ của sự suy giảm kali bị ẩn khuất do các xu hướng toan chuyển hóa máu để thúc đẩy kali ra khỏi tế bào. Như vậy, nồng độ kali huyết thanh cao hơn nên được liên quan đến mức độ của sự suy giảm kali. Ở một số bệnh nhân, nồng độ kali huyết thanh bình thường hoặc thậm chí cao, mặc dù sự điều chỉnh của toan chuyển hóa máu sẽ phát hiện tình trạng thực sự của cân bằng kali.
Trước khi điều trị với bicarbonate, bệnh nhân có nhiễm toan ống thận gần thường chứng minh hạ kali máu nhẹ, chủ yếu là do cường aldosteron cơ bản. Tuy nhiên, điều trị bằng natri bicarbonate để sửa toan chuyển hóa máu gây tăng natri ở ống xa và cung cấp bicarbonate, có thể gây ra một sự gia tăng đáng kể tiêu hao kali thận.
Giảm magiê máu — Giảm magiê máu biểu hiện ở > 40% các bệnh nhân giảm kali máu. Ở nhiều trường hợp, như với điều trị lợi tiểu, nôn ói, tiêu chảy, hoặc nhiễm độc ống thận như gentamicin và iphosphamide, có mất đồng thời kali và magiê.
Tuy nhiên giảm magiê máu có thể tự bản thân nó đưa đến mất kali niệu tăng lên do cơ chế còn chưa rõ, có khả năng liên quan đến tăng số các kệnh kali mở Cơ chế này, cũng như các nguyên nhân giảm magiê máu là một chuyên để lớn.
Xác định có giảm magiê hay không là một vấn đề lâm sàng quan trọng vì giảm kali thường không được sửa chữa đến tận khi magiê thiếu hụt được phục hồi.
Amphotericin B — Giảm kali máu xuất hiện ở trên một nửa bệnh nhân được điều trị bằngamphotericin B. Amphotericin tương tác với sterole màng tế bào, đưa đến tăng tính thấm màng tế bào có thể thúc đẩy tiết kali qua màng tế bào trong lòng ống. Nhiễm toan ống thận đầu xa có thể đòng vai trò phụ thêm.
Các bệnh thận gây tiêu hao muối (Salt-wasting nephropathies) — Các bệnh thận kết hợp với tái hấp thu natri ở đầu gần, quai, đầu xa giảm xuống có thể đưa đến giảm kali do cơ chế tương tự được gây ra do lợi tiểu ít gặp hơn. Vấn đề này có thể được thấy trong hội chứng Bartter hoặc Gitelman, các bệnh ống kẽ thận (chẳng hạn các bệnh thận ngược dòng hoặc viêm thận kẽ do hội chứng Sjögren), tăng canxi máu, tổn thương ống thận do thuốc như cisplatin, và tổn thương ống thận có thể được tạo ra do lysozyme ở các bệnh nhân bệnh bạch cầu, đặc biệt bạch cầu dòng mono hoặc mono dòng tủy)
Hội chứng Liddle — Ở các bệnh nhân hội chứng Liddle, autosom chiếm ưu thế, đột biến gain chức năng trong Ena C đưa đến giảm kali di truyền và tăng huyết áp, giống hệt như hội chứng tiết hormone khoáng (mineralocorticoid).
Hội chứngBartter và Gitelman — Các đột biến đa dạng của các proteins vận chuyển của ống thận có thể giống hệt ảnh hưởng của điều trị kéo dài các thuốc lợi tiểu quai (hội chứng Bartter) hoặc lợi tiểu thiazide (hội chứng Gitelman), dẫn đến hạ kali máu và nhiễm kiềm chuyển hóa.
Chế độ ăn ít calo – Ăn một chế độ ăn ít calo (200 và 800 kcal/ngày) có thể dẫn đến hạ kali máu, đặc biệt ở những bệnh nhân không uống kali bổ sung hoặc những người có khuynh hướng tiêu hao kali thận cơ bản như cường aldosterone tiên phát ở nhiều bệnh nhân lúc đầu kali bình thường. Những chế độ ăn kiêng, dùng để tạo ra giảm cân nhanh chóng, thường là nghèo carbohydrate và protein phong phú, chẳng hạn như chế độ ăn Atkins.
Cơ chế của hạ kali ở các bệnh nhân ăn chế độ ít calo không hoàn toàn được hiểu rõ. Tiêu hao kali đường niệu được cho là quan trọng do các bệnh nhân béo phì chứng tỏ kali niệu có ý nghĩa qua hai tuần đầu tiên ăn chay hoặc ăn chế độ carbohydrate thấp. Kali niệu có thể do ít nhất một phần đưa đến tạo cetone do giảm nhập carbohydrate. Như đã nói trên, tiết các anions axit ceton có thể tăng tiết kali niệu.
MẤT DO TĂNG TIẾT MỒ HÔI — Mất kali qua mồ hôi hàng ngày là bình thường không đáng kể do khối lượng thấp và nồng độ kali là chỉ 5-10 mEq/l. Tuy nhiên, các cá nhân gắng sức trong khí hậu nóng bức có thể tiết 10 L mô hôi hoặc nhiều hơn mỗi ngày, dẫn đến sự suy giảm kali nếu những mất mát không được bù. mất kali đáng kể trong mồ hôi cũng có thể xảy ra ở bệnh nhân xơ nang (cyslic fibrosis). Nước tiểu bài tiết kali có thể góp phần vào việc giảm kali máu kết hợp với tăng mất qua mồ hôi từ đó tiết aldosterone được gia tăng do gắng sức (qua việc tiết renin được tạo ra do catecholamine).
Lọc máu – Mặc dù bệnh nhân có bệnh thận giai đoạn cuối thường giữ lại kali và có xu hướng tăng kali máu nhẹ, hạ kali máu có thể gây ra ở một số bệnh nhân chạy thận duy trì. Như ví dụ, chạy thận mất kali có thể đạt 30 mEq mỗi ngày ở những bệnh nhân thẩm phân phúc mạc kéo dài. Điều này có thể trở thành vấn đề lâm sàng quan trọng nếu lượng kali trong ăn uống giảm hoặc nếu có những mất theo đường tiêu hóa đồng thời. Tuy nhiên, hạ kali máu nhẹ ngay sau chạy thận nhân tạo được mong đợi và không nên điều trị bằng bổ sung kali.
Cơ chế khác có thể mở ra ở những bệnh nhân được điều trị bằng chạy thận nhân tạo có nền giảm kali. Trong tình trạng này, toan chuyển hóa do suy thận có thể dẫn đến nồng độ kali máu trước lọc máu tương đối bình thường do sự chuyển động kali ra khỏi tế bào. Tuy nhiên, các lưu lượng cao đạt được trong quá trình chạy thận nhân tạo có thể nhanh chóng sửa toan chuyển hóa máu, dẫn đến nhập kali vào tế bào và khả năng giảm kali máu nặng.
Huyết tương – Huyết tương loại bỏ kali ở nồng độ tương tự như trong huyết tương. Tuy nhiên, nếu albumin được sử dụng như là chất lỏng thay thế, hạ kali máu thoáng qua có thể được gây ra do sự pha loãng. Vấn đề này có thể tránh được bằng cách thêm 4 mEq kali cho mỗi lít dung dịch albumin nhất định.
Tài liệu tham khảo
1. SQUIRES RD, HUTH EJ. Experimental potassium depletion in normal human subjects. I. Relation of ionic intakes to the renal conservation of potassium. J Clin Invest 1959; 38:1134. 2. Mount DB, Zandi-Nejad K. Disorders of potassium balance. In: Brenner and Rector’s The Kidney, 8th ed, Brenner BM (Ed), WB Saunders Co, Philadelphia 2008. p.547. 3. Clausen T. Hormonal and pharmacological modification of plasma potassium homeostasis. Fundam Clin Pharmacol 2010; 24:595. 4. Fuentebella J, Kerner JA. Refeeding syndrome. Pediatr Clin North Am 2009; 56:1201. 5. Gosmanov AR, Wong JA, Thomason DB. Duality of G protein-coupled mechanisms for beta-adrenergic activation of NKCC activity in skeletal muscle. Am J Physiol Cell Physiol 2002; 283:C1025. 6. Williams ME, Gervino EV, Rosa RM, et al. Catecholamine modulation of rapid potassium shifts during exercise. N Engl J Med 1985; 312:823. 7. Shannon M, Lovejoy FH Jr. Hypokalemia after theophylline intoxication. The effects of acute vs chronic poisoning. Arch Intern Med 1989; 149:2725. 8. Wijkerslooth LR, Koch BC, Malingré MM, et al. Life-threatening hypokalaemia and lactate accumulation after autointoxication with Stacker 2, a ‘powerful slimming agent’. Br J Clin Pharmacol 2008; 66:728. 9. Lin SH, Lin YF, Chen DT, et al. Laboratory tests to determine the cause of hypokalemia and paralysis. Arch Intern Med 2004; 164:1561. 10. Rose BD, Post TW. Clinical Physiology of Acid-Base and Electrolyte Disorders, 5th ed, McGraw-Hill, New York 2001. p.836. 11. Schaller MD, Fischer AP, Perret CH. Hyperkalemia. A prognostic factor during acute severe hypothermia. JAMA 1990; 264:1842. 12. Sigue G, Gamble L, Pelitere M, et al. From profound hypokalemia to life-threatening hyperkalemia: a case of barium sulfide poisoning. Arch Intern Med 2000; 160:548. 13. Wells JA, Wood KE. Acute barium poisoning treated with hemodialysis. Am J Emerg Med 2001; 19:175. 14. Melnikov P, Zanoni LZ. Clinical effects of cesium intake. Biol Trace Elem Res 2010; 135:1. 15. Malik AR, Wolf PK, Ravasia S. Hypokalemia from risperidone and quetiapine overdose. Can J Psychiatry 2005; 50:76. 16. Lin YC, Chen HZ, Chang TJ, Lane HY. Hypokalemia following rapid titration of quetiapine treatment. J Clin Psychiatry 2008; 69:165. 17. Older J, Older P, Colker J, Brown R. Secretory villous adenomas that cause depletion syndrome. Arch Intern Med 1999; 159:879. 18. Sitprija V. Altered fluid, electrolyte and mineral status in tropical disease, with an emphasis on malaria and leptospirosis. Nat Clin Pract Nephrol 2008; 4:91. 19. Rose BD, Post TW. Clinical Physiology of Acid-Base and Electrolyte Disorders, 5th ed, McGraw-Hill, New York 2001. p.333. 20. Ho JM, Juurlink DN, Cavalcanti RB. Hypokalemia following polyethylene glycol-based bowel preparation for colonoscopy in older hospitalized patients with significant comorbidities. Ann Pharmacother 2010; 44:466. 21. Beloosesky Y, Grinblat J, Weiss A, et al. Electrolyte disorders following oral sodium phosphate administration for bowel cleansing in elderly patients. Arch Intern Med 2003; 163:803. 22. Bennett A, Stryjewski G. Severe hypokalemia caused by oral and rectal administration of bentonite in a pediatric patient. Pediatr Emerg Care 2006; 22:500. 23. Ukaonu C, Hill DA, Christensen F. Hypokalemic myopathy in pregnancy caused by clay ingestion. Obstet Gynecol 2003; 102:1169. 24. Wang WH, Giebisch G. Regulation of potassium (K) handling in the renal collecting duct. Pflugers Arch 2009; 458:157. 25. Welling PA, Chang YP, Delpire E, Wade JB. Multigene kinase network, kidney transport, and salt in essential hypertension. Kidney Int 2010; 77:1063. 26. Vallon V, Schroth J, Lang F, et al. Expression and phosphorylation of the Na+-Cl- cotransporter NCC in vivo is regulated by dietary salt, potassium, and SGK1. Am J Physiol Renal Physiol 2009; 297:F704. 27. Wang ZJ, Sun P, Xing W, et al. Decrease in dietary K intake stimulates the generation of superoxide anions in the kidney and inhibits K secretory channels in the CCD. Am J Physiol Renal Physiol 2010; 298:F1515. 28. Babilonia E, Wei Y, Sterling H, et al. Superoxide anions are involved in mediating the effect of low K intake on c-Src expression and renal K secretion in the cortical collecting duct. J Biol Chem 2005; 280:10790. 29. Papademetriou V. Diuretics, hypokalemia, and cardiac arrhythmia: a 20-year controversy. J Clin Hypertens (Greenwich) 2006; 8:86. 30. Schmieder RE, Rockstroh JK. Efficacy and tolerance of low-dose loop diuretics in hypertension. Cardiology 1994; 84 Suppl 2:36. 31. Carlsen JE, Køber L, Torp-Pedersen C, Johansen P. Relation between dose of bendrofluazide, antihypertensive effect, and adverse biochemical effects. BMJ 1990; 300:975. 32. ALLHAT Officers and Coordinators for the ALLHAT Collaborative Research Group. The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial. Major outcomes in high-risk hypertensive patients randomized to angiotensin-converting enzyme inhibitor or calcium channel blocker vs diuretic: The Antihypertensive and Lipid-Lowering Treatment to Prevent Heart Attack Trial (ALLHAT). JAMA 2002; 288:2981. 33. Franse LV, Pahor M, Di Bari M, et al. Hypokalemia associated with diuretic use and cardiovascular events in the Systolic Hypertension in the Elderly Program. Hypertension 2000; 35:1025. 34. Mohr JA, Clark RM, Waack TC, Whang R. Nafcillin-associated hypokalemia. JAMA 1979; 242:544. 35. Carlisle EJ, Donnelly SM, Vasuvattakul S, et al. Glue-sniffing and distal renal tubular acidosis: sticking to the facts. J Am Soc Nephrol 1991; 1:1019. 36. Hariprasad MK, Eisinger RP, Nadler IM, et al. Hyponatremia in psychogenic polydipsia. Arch Intern Med 1980; 140:1639. 37. Husband DJ, Watkin SW. Fatal hypokalaemia associated with ifosfamide/mesna chemotherapy. Lancet 1988; 1:1116. 38. Huang CL, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol 2007; 18:2649. 39. Yang L, Frindt G, Palmer LG. Magnesium modulates ROMK channel-mediated potassium secretion. J Am Soc Nephrol 2010; 21:2109. 40. Perazella MA, Eisen RN, Frederick WG, Brown E. Renal failure and severe hypokalemia associated with acute myelomonocytic leukemia. Am J Kidney Dis 1993; 22:462. 41. Mir MA, Brabin B, Tang OT, et al. Hypokalaemia in acute myeloid leukaemia. Ann Intern Med 1975; 82:54. 42. Aldinger KA, Samaan NA. Hypokalemia with hypercalcemia. Prevalence and significance in treatment. Ann Intern Med 1977; 87:571. 43. Liu T, Nagami GT, Everett ML, Levine BS. Very low calorie diets and hypokalaemia: the importance of ammonium excretion. Nephrol Dial Transplant 2005; 20:642. 44. Singh BN, Gaarder TD, Kanegae T, et al. Liquid protein diets and torsade de pointes. JAMA 1978; 240:115. 45. Advani A, Taylor R. Life-threatening hypokalaemia on a low-carbohydrate diet associated with previously undiagnosed primary hyperaldosteronism [corrected]. Diabet Med 2005; 22:1605. 46. Rabast U, Vornberger KH, Ehl M. Loss of weight, sodium and water in obese persons consuming a high- or low-carbohydrate diet. Ann Nutr Metab 1981; 25:34. 47. Davé S, Honney S, Raymond J, Flume PA. An unusual presentation of cystic fibrosis in an adult. Am J Kidney Dis 2005; 45: 41.
Từ khóa » Tốc độ Truyền Kali
-
Nguyên Tắc Sử Dụng KCl đường Tĩnh Mạch
-
Hạ Kali Máu - HSCC
-
Hạ Kali Máu - Rối Loạn Nội Tiết Và Chuyển Hóa - MSD Manuals
-
ANSM: Nhắc Lại Nguyên Tắc Trong Sử Dụng Kali Clorua đường Tĩnh Mạch
-
[PDF] RỐI LOẠN KALI MÁU - UMP
-
Dược Thư - Y Khoa Phước An
-
Hạ Kali Máu - PGS Hà Hoàng Kiệm
-
Hạ Kali Máu – Tiếp Cận Chẩn đoán Và điều Trị
-
Hạ Kali Máu: Chẩn đoán Và điều Trị
-
[PDF] HẠ KALI MÁU - Bệnh Viện Tim Mạch An Giang
-
HẠ KALI MÁU
-
Kali Clorid (Potassium Chloride): Công Dụng Và Lưu ý Khi Dùng
-
Phác đồ điều Trị Cấp Cứu Hạ Kali Máu
-
RỐI LOẠN NATRI VÀ KALI MÁU - Health Việt Nam
-
Vì Sao Thuốc Kali Clorid Dễ Bị Nhầm Lẫn?
-
Điều Trị Và Xử Trí Hạ Kali Máu | Vinmec
-
[PDF] HƯỚNG DẪN SỬ DỤNG MỘT SỐ LOẠI THUỐC TIÊM, TIÊM TRUYỀN
-
Compound Sodium Lactate Braun | BvNTP
-
Tăng Kali Máu | BvNTP